β-5型木脂素气化反应机理的密度泛函理论
2017-10-16邓胜祥曹小玲
张 航, 邓胜祥, 田 红, 曹小玲
(1.中南大学 能源科学与工程学院,湖南 长沙 410083; 2.长沙理工大学 能源与动力工程学院, 湖南 长沙 410015)
β-5型木脂素气化反应机理的密度泛函理论
张 航1, 邓胜祥1, 田 红2, 曹小玲2
(1.中南大学 能源科学与工程学院,湖南 长沙 410083; 2.长沙理工大学 能源与动力工程学院, 湖南 长沙 410015)
在密度泛函理论的基础上,采用B3LYP/6-31+G(d,p)方法对β-5型木脂素的气化反应机理进行研究,考察温度对气化反应机理的影响。设计3条热解反应路径及相应的后继反应,并对反应路径中各驻点结构进行能量梯度全优化以及振动频率计算。热力学计算结果表明,在中低温(T<1500 K)阶段,Path 1、4、5、6和7的焓变(ΔHθ)均大于0,反应是吸热的;而Path 2的ΔHθ在温度达到1500 K后降为-50.6 kJ/mol,反应从低温时吸热变为高温时放热;Path 3的焓值从2.1 kJ/mol逐渐减小到-125.1 kJ/mol,反应由吸热变为放热。当300 K
木脂素; 密度泛函理论; 气化; 热力学; 动力学
Abstract: Gasification of the lignan model compound withβ-5 linkage was theoretically investigated by employing density functional theory methods at B3LYP/6-31G+(d,p) level. Three pyrolysis reaction pathways and corresponding subsequent reactions were considered. The equilibrium geometries of the reactants, transition states, intermediates and products were optimized, and the standard kinetic parameters of reaction pathways were calculated. Thermodynamics calculation results showed that enthalpy changes of Path 1, 4, 5, 6 and 7 were greater than 0 at the low temperature (T<1500 K), and the reactions were endothermic. The enthalpy change of Path 2 decreased to -50.6 kJ/mol when temperature reached to 1500 K. The enthalpy of Path 3 decreased from 2.1 kJ/mol to -125.1 kJ/mol, and the reaction changed from endothermic to exothermic. Path 2 and Path 3 could occur spontaneously at temperature arrange from 300 K to 1700 K. WhenTwas larger than 700 K, the Path 1 could also occur spontaneously, but Path 4, 5, 6 and 7 couldn’t. Dynamics calculation results showed that first step of Path 1, 2, 3, the corresponding reaction activation energies were 192.5, 159.0 and 113.0 kJ/mol, respectively. The second step reaction activation energies were 138.1, 184.1 and 280.3 kJ/mol, respectively. The third step of Path 1 reaction was 79.5 kJ/mol. Path 2 was the optimal reaction path, namely Cα—Cβkey fractured more easily in the lignan model compound. Path 4 was optimal in the subsequent reaction. The main products of Path 2 were (2-hydroxyphenyl) acetaldehyde and 4-(hydroxymethyl) phenol. In the subsequent reactions, the products were CO, CO2, H2, phenol and methanol.
Keywords:lignan model compound; density functional theory; gasification; thermodynamics; dynamics
当今世界能源和环境问题日益突出,急需可再生、储量大的清洁能源来代替化石能源,所以生物质能及其利用技术逐渐引起人们的关注[1]。生物质高温蒸汽气化技术作为一种极具前景的生物质利用技术,具有NOx排放的体积分数低于10-4、节能15%~30%和CO2减排量达30%的优点[2-3]。而木质素是生物质的三大组分之一,它的气化特性在很大程度上可以体现出生物质整体的气化规律,因此有必要对其气化过程进行微观反应机理研究[4-5]。木质素是通过单体脱氢聚合,由C—C键和C—O键等连接无序组合而成[6-7]。而β-5型木脂素(见图1)是木质素中的一种典型结构[8],主要由2分子的苯丙素衍生物构成,分子间通过β位的碳连接(β-5型表示β位的碳和5号位的碳相连)。因此β-5型木脂素能很好地诠释木质素气化机理。

图1 β-5型木脂素分子结构示意图Fig.1 Molecular structure schematic of lignan modelcompound with β-5 linkage
谭洪等[9]和岳金方等[10]在木质素热解实验的产物中发现大量的苯酚类物质。Qin等[11]在生物质气化实验中发现,富含木质素的锯末的气化产物焦油中含有大量带侧链的芳香族化合物,随着温度升高,侧链部分裂解。刘江燕[12]同样也在实验产物中发现苯酚类化合物,并推断木质素在热解过程中C—Cβ发生断裂,因为只有Cα—Cβ和Cβ—C5断裂才会产生苯酚类物质。王华静等[13]的实验结果同样证明,木质素二聚体裂解的主要产物,Cα—Cβ键断裂的反应路径以及β-O-4键断裂反应路径中的主要产物都是苯酚、对羟基苯甲醇和乙醇。学者们的研究大多集中在β-O-4型木质素,关于该型木质素的热裂解机理已有诸多论述。Elder等[14]对β-O-4型木质素模化物的热解机理进行量子化学研究,计算得出协同反应的活化能低于均裂反应的键离解能,表明均裂反应占主导地位。而黄金保等[15]认为,均裂反应和协同反应都是木质素二聚体的主要反应路径,二者之间的选择受温度的影响,在低温时协同反应是热解过程中的主要反应形式,而高温阶段自由基均裂反应是主要反应形式。除β-O-4型还有其他键型的木质素,如Huang等[16]曾探索β-1型木质素的热裂解机理。武书彬等[17]对α-O-4型木质素二聚体的热裂解行为进行研究。曹小玲等[18]在水溶剂环境下研究β-1型木质素二聚体的高温蒸汽气化反应机理。关于木质素在高温蒸汽气化过程中发生的化学反应,以及主要产物的生成机理、中间体和过渡态的演变反应均少有报道。
笔者采用密度泛函理论的B3LYP方法和6-31+G(d,p)基组对β-5型木脂素在不同温度的纯蒸汽气化反应进行计算,从热力学和动力学角度对计算结果进行分析。
1 β-5型木脂素气化反应路径
根据分子轨道理论,前沿轨道(分为最高占据轨道(HOMO)和最低未占轨道(LUMO))及其附近的分子轨道对物质反应活性影响最大,HOMO及附近的占据轨道具有优先提供电子的作用,LUMO及附近的空轨道具有接受电子的重要作用[19-20]。亲电反应最易发生在HOMO最大电荷密度的原子上;与此类似,亲核反应在各个原子上发生的相对次序由LUMO的电荷密度分布决定,亲核试剂最易进攻LUMO电荷密度最大的原子。β-5型木脂素的气化反应应当发生在前沿轨道中各基团电子云密度最大的某个原子上。HOMO的能量在一定程度上反映了分子得失电子的难易程度,从而可以分析其参与反应的难易程度。表1为应用Gaussian09商业软件并采用密度泛函理论计算得到的β-5型木脂素分子的HOMO能量和原子电荷密度。

表1 β-5型木脂素分子的前沿轨道HOMO能量(E)和原子电荷密度Table 1 Frontier orbital analysis and atomic charge density of the lignan with β-5 linkage
由表1可知,C3和C10原子部位的电荷密度比其他原子的电荷密度小1个数量级,所以这个原子部位很难发生反应,即苯环难以发生反应,这也解释了木质素裂解实验产物中存在大量含有苯基化合物的原因[9]。而Cα—Cβ、Cγ—Cβ、Cβ—C5、C4—OH、C9—OH等基团位置的原子电荷密度相对较大,所以反应就容易发生在这些苯环相连的原子部位上。由于C4和C9上的羟基分别属于2个苯环的侧链基团,它们的反应势垒非常大[18],因此不考虑该位置的反应路径。β-5型木脂素的气化反应是双苯环主体中间的碳链在外界环境变化的作用下发生断裂生成单苯环带侧链的基团。据此,笔者设计多条反应途径来描述上述反应过程,经过归纳和总结,反应路径分别表示为Cβ—C5、Cα—Cβ和Cγ—Cβ键的均裂及其后继反应,如图2所示。
Path 1的具体过程:Cβ—C5均裂,其中Cα失去H后与Cβ形成双键,得到化合物IM1,而C5吸附H后得到产物P3;IM1的Cγ—Cβ均裂,其中Cγ失去H演变为甲醛,Cβ吸收H后,得到IM4;IM4的异构化反应,Cα的氢氧基上的H转移到Cβ,出现过渡态TS7,继而Cα与相连的O组成醛基,生成苯甲醛。
Path 2的具体过程: Cα—Cβ均裂,其中Cγ失去H后与Cβ形成双键,得到中间体IM2,而Cα吸附H生成产物P4;IM2发生异构化反应,Cγ—OH上的H被Cβ得到,Cγ与相连的O组成醛基,生成P5。
Path 3的具体过程:Cγ—Cβ均裂,其中Cγ—OH 发生脱氢反应,Cγ与O组成醛基,形成产物P1,而Cβ吸附H后得到IM3;IM3的Cα—Cβ断开,Cα—OH上的H被Cβ得到,失去H的—OH与Cα形成醛基,得到产物P6和P7。
将典型产物P2、P4、P5和P6作为后继反应的反应物,继续和水蒸气发生反应。笔者设计4条后继反应路径,见图3。
2 密度泛函理论计算方法
密度泛函理论(DFT)在考虑电子相关效应的同时,不仅可进行大分子体系的计算,而且计算结果准确、可靠[21]。对于较轻的元素,如C、H、O、N等,适合采用B3LYP/6-31G计算方法[22-23]。笔者采用DFT,并在B3LYP/6-31+G(d,p)水平上对路径中各驻点(反应物、中间体、产物)以及过渡态的几何构型进行优化。B3LYP是指Becke型3参数密度泛函与Lee-Yang-Parr泛函的混合泛函。6-31G是描述原子的基组,6-31+G(d,p)是在6-31G的基础上对第1周期的H原子和第2周期的C原子添加极化基函数,而极化基函数的极化基组比原基组能更好地描述体系。笔者在不同温度(T为300、500、700、900、1100、1300、1500、1700 K)下计算各驻点的振动频率,通过振动分析并确认每1个驻点都无虚频,获取热力学参数。采用Berny优化(相应的关键词为OPT=TS)方法在同一水平下寻找过渡态,计算过渡态的振动频率,并通过振动频率和内禀反应坐标分析(IRC)方法对过渡态的真实性进行验证[24]。
3 结果与讨论
3.1β-5型木脂素气化反应中部分反应物、中间体、产物和过渡态的几何构型优化
表2为结构优化后的反应物、中间体、产物和过渡态的几何结构及关键键长。
由表2可知,反应物、中间体和产物都无虚频存在,而所得过渡态有且只有唯一的虚频,说明优化得到的各分子构型是合理的。过渡态的虚频见表3。

图2 β-5型木脂素的热解反应路径示意图Fig.2 Pyrolysis reaction path schematic of the lignan with β-5 linkage

图3 β-5型木脂素气化的后继反应路径示意图Fig.3 Subsequent reaction path schematic of the lignan with β-5 linkage

OptimizedstructureStructuralparameterBondlength/(0.1nm)Bondangle/(°)Dihedral/(°)R(1,2)1.4132A(2,1,6)117.1112D(6,1,2,3)-0.5293R(1,6)1.3997A(2,1,20)122.5438D(6,1,2,31)179.1842R(1,20)1.5060A(6,1,20)120.3416D(20,1,2,3)178.8091R(2,3)1.4003A(1,2,3)122.5626D(20,1,2,31)-1.4774R(2,31)1.3807A(1,2,31)116.3465D(2,1,6,5)0.4037R(1,2)1.4029A(2,1,6)122.3725D(6,1,2,3)-0.1713R(1,6)1.4065A(2,1,21)122.5007D(6,1,2,7)179.7040R(1,21)1.3720A(6,1,21)115.1268D(21,1,2,3)179.9118R(2,3)1.3948A(1,2,3)118.2303D(21,1,2,7)-0.2129R(2,7)1.0847A(1,2,7)121.2197D(2,1,6,5)-0.1716R(1,2)1.0871A(2,1,3)118.0958D(6,1,2,3)-0.4612R(1,3)1.4498A(2,1,8)121.9716D(6,1,2,11)179.9073R(1,8)1.3471A(3,1,8)119.9300D(15,1,2,3)179.4959R(3,4)1.0879A(1,3,4)119.4839D(15,1,2,11)-0.1356R(3,5)1.3489A(1,3,5)118.2781D(2,1,6,5)0.1786R(1,2)1.4211A(2,1,6)117.4823D(6,1,2,3)-0.2929R(1,6)1.3977A(2,1,20)120.4879D(6,1,2,26)179.4987R(1,15)1.3786A(6,1,20)122.0280D(20,1,2,3)179.7536R(2,3)1.4009A(1,2,3)122.4927D(20,1,2,26)-0.4548R(2,11)1.4576A(1,2,26)115.5588D(2,1,6,5)0.1704

续表2
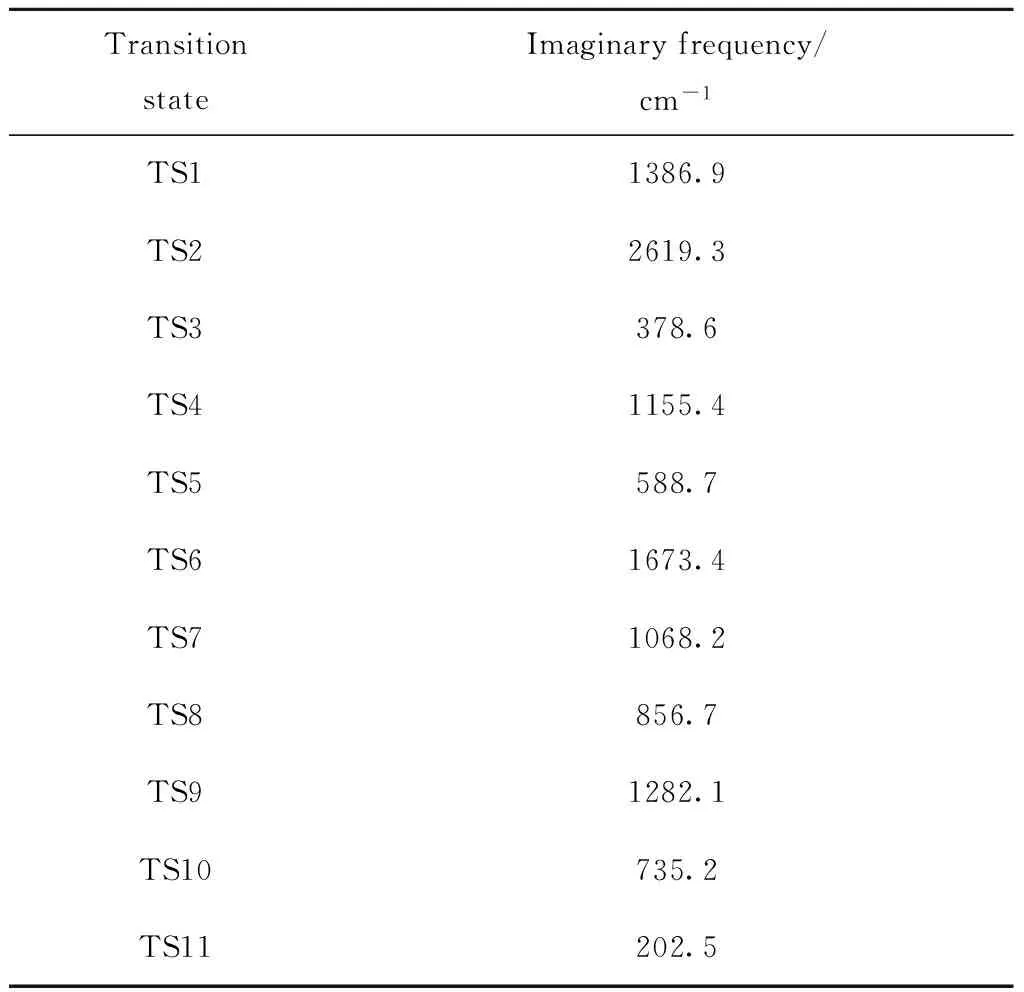
表3 β-5型木脂素气化反应中过渡态的虚频Table 3 Imaginary frequency of the transition states forthe β-5 lignan gasification reaction
3.2β-5型木脂素气化反应的热力学分析
通过各驻点几何构型优化后的热力学数据,即不同温度下的反应焓变(ΔHθ)和吉布斯自由能变(ΔGθ),来判断各反应路径发生的优先顺序以及活跃程度。当ΔGθ<0时,反应可自发进行,ΔGθ越小,反应达到平衡时,反应物的转化率越大;当ΔHθ<0时,反应为放热反应,反之则为吸热反应。
笔者在不同温度的水蒸气环境下计算出β-5型木脂素气化反应路径的焓变和吉布斯自由能变。图4 为β-5型木脂素气化反应的焓变随温度的变化曲线。由图4(a)看到,随着温度升高,各反应的ΔHθ减小,且减小量逐渐增大。当温度T=300 K时,Path 1的ΔHθ为173.6 kJ/mol,随着温度提高,ΔHθ缓慢减小但始终大于0,所以Path 1 始终是吸热反应;当T=300 K时,Path 2的吸热量为32.2 kJ/mol,ΔHθ也随着温度的升高而变小,但温度升到1500 K时,ΔHθ<0,反应变为放热;当T=300 K时,Path 3的ΔHθ>0,仅有2.1 kJ/mol,当温度升高到500 K时,反应就开始放热,且放热量随温度升高也越来越大,当温度达到1700 K时,放热量为125.1 kJ/mol。
由图4(b)看到,当T在300~1700 K范围时,Path 4的ΔHθ>0,且吸热量随温度升高而减小,当温度达到1700 K时,吸热量仅为6.7 kJ/mol;当T=300 K时,Path 5的ΔHθ=61.9 kJ/mol,当温度升高到1500 K以上,反应由吸热转到放热,当温度达到1700 K时放热量为4.6 kJ/mol;当T=300 K时,Path 6的ΔHθ=84.9 kJ/mol,且吸热量随温度升高而减小,当温度达到1700 K时,吸热量仅为8.4 kJ/mol;当T=300 K时,Path 7的ΔHθ=53.6 kJ/mol,当温度升高到1500 K以上,反应逐渐变为放热,当温度达到1700 K时放热量为3.3 kJ/mol。
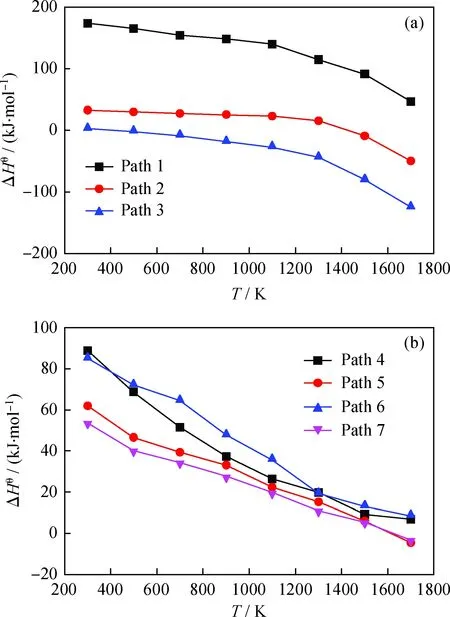
图4 β-5型木脂素气化反应的焓变(ΔHθ)随温度(T)的变化曲线Fig.4 Enthalpy changes (ΔHθ) vs temperature (T) forthe β-5 lignan gasification reaction (a) Pyrolysis reaction path; (b) Subsequent reaction path
图5为β-5型木脂素气化反应的吉布斯自由能变随温度变化的曲线。由图5(a)看到,当T在300~1700 K范围时,Path 2和Path 3的ΔGθ均小于0,即Path 2、Path 3都能自发进行。当T>700 K时,Path 1的ΔGθ<0,所以Path 1也可以自发进行。当T在300~1100 K范围时,ΔGθ排列由小到大的顺序为Path 3、Path 2、Path 1,而当T在1100~1700 K时,ΔGθ排列由小到大的顺序为Path 3、Path 1、Path 2。由图5(b)看到,后继反应路径的ΔGθ计算结果表明,当T在300~1700 K范围时,Path 4、5、6、7的ΔGθ均大于0,即所有后继反应都不能自发进行。

图5 β-5型木脂素气化反应的吉布斯自由能变(ΔGθ)随温度(T)变化的曲线Fig.5 Gibbs free energy changes (ΔGθ) vstemperature (T) for the β-5 lignan gasification reaction (a) Pyrolysis reaction path; (b) Subsequent reaction path
3.3β-5型木脂素气化反应的动力学分析
根据过渡态理论[25-26],活化能即是位能面上过渡态与反应物都处于基态时的最低位能差。由于热力学变量随温度的变化非常小,所以通常用总能量(E0)来代替反应的活化能。通过计算[24]得到各构象的总能量见表4。
图6为图2中β-5型木脂素热解反应路径的势能剖面图。从图6可以看出,Path 1的反应物发生均裂反应。第1步,C5—Cβ键变长拉断,然后形成过渡态TS1a和TS1b,其中TS1b的C5从Cβ处吸引1个H原子,Cα与Cβ以双键连接并产生稳定的中间体IM1,而TS1a直接得到产物P3。该反应步的活化能为192.5 kJ/mol。反应继续进行,随着能量的累积,Cγ—Cβ键变长后断开,其中Cγ失去H原子后与O原子形成Cγ=O,而Cβ吸收1个H原子后趋于稳定状态。反应倾向于生成甲醛和IM4,该步的活化能为138.1 kJ/mol。第3步是IM4的异构化反应,Cα—OH上的H原子转移到Cβ,而Cα与相连的O原子组成醛基,最终产物是P2,这步反应的活化能只有79.5 kJ/mol。因此在这条反应路径上,第3步是决速步。

表4 β-5型木脂素气化反应中各构型的总能量(E0)Table 4 The total energy (E0) of species for the β-5 lignan gasification reaction
Path 2只有2步反应。第1步,反应物的Cα—Cβ键拉长断裂,其中Cβ失去1个H原子后与Cγ形成Cβ=Cγ,而Cα吸引1个H原子。该反应步的产物为IM2和P4,活化能为159.0 kJ/mol。第2步,IM2的异构化反应,Cγ的—OH脱离1个H原子,然后组成Cγ=O,而Cβ吸引1个H原子,得到产物P5。此步反应的活化能为184.1 kJ/mol。第1步是Path 2的决速步。
Path 3也只有2步反应。首先Cγ—Cβ键断,Cβ吸收1个H原子后得到中间体IM3,而Cγ与O形成双键,生成甲醛,反应活化能为113.0 kJ/mol。第2步, Cα—OH脱氢组成Cα=O,而Cβ得到H后生成产物P7,该步反应的活化能为280.3 kJ/mol。同样,第1步是这条反应路径的决速步。
横向比较各路径对应的反应活化能,第1步中Path 3的活化能最低,第2步的活化能最低的是Path 2。由于只有Path 1存在第3步,因此也是难以发生的反应路径。而Path 2的综合活化能要低于Path 3,所以动力学分析支持Path 2为最优反应路径。综上所述,β-5型木脂素的Cβ—Cα键更易断裂。

图6 图2中β-5型木脂素热解反应途径的势能剖面图Fig.6 Potential energy profile of the β-5 lignan pyrolysisreaction paths in Fig.2S1=P3+IM1; S2=P1+IM3; S3=P4+IM2; S4=P1+ P6+P7;S5=P3+ P1+IM4; S6=P4+ P5; S7=P3+ P1+P2
图7为图3中后继反应路径的势能剖面图。由图7可见,Path 4的具体过程:产物P2的支链与主体分离,同时H—OH断开,苯环得到1个H原子生成苯酚(P3),而支链上的Cα—Cβ均裂,Cβ得到—OH生成甲醇,Cα与O组成CO,该反应的活化能为66.9 kJ/mol。Path 5的具体过程:Cα—C1断裂,同时H—OH断开,C1得到H原子生成苯酚(P3),而Cα得到—OH后生成CO2和2个H2,该反应的活化能为171.5 kJ/mol。Path 6的具体过程:C5—Cβ断裂,同时H—OH断开,C5得到1个H原子,而Cβ与Cγ发生均裂反应,Cβ得到—OH生成甲醇,Cγ与O组成双键生成CO,该反应的活化能为75.3 kJ/mol。Path 7的具体过程:Cα—C1断裂,C1得到1个H原子生成苯酚(P3),而Cα得到—OH生成产物CO2和H2,该反应的活化能为92.0 kJ/mol。对比各反应路径的活化能,Path 4的活化能相对较小,其次是Path 6,最大也是最难发生的是Path 5。所以动力学分析支持Path 4为最优反应路径。

图7 图3中β-5型木脂素后继反应途径的势能剖面图Fig.7 Potential energy profile of the β-5 lignansubsequent reaction paths in Fig.3
4 结 论
采用密度泛函理论B3LYP/6-31+G(d,p)对β-5 型木脂素在不同温度的蒸汽气化反应机理和产物形成进行研究。提出3条热解反应路径以及后继反应,并计算路径中各驻点的活化能。得出主要结论如下:
(1)计算反应路径的焓变表明,Path 2在温度达到1500 K后变为放热反应,而Path 3在温度超过500 K就是放热反应。其他热解反应路径和后继反应在中低温(T<1500 K)时为吸热反应。吉布斯自由能变的计算结果表明,在设计温度内,Path 2、Path 3都能自发进行;当T>700 K时,Path 1也可以自发进行,而Path 4、5、6和7均不能自发进行。
(2) 3条热解反应路径的第1步反应活化能由小到大的顺序为Path 3、Path 2、Path 1,而第2步反应的活化能由小到大的顺序为Path 2、Path 1、Path 3,综合比较活化能,则Path 2为最优反应路径,所以β-5型木脂素的Cβ—Cα键更容易断裂。后继反应中, Path 4的反应活化能相对最小,所以反应最容易进行。β-5型木脂素热解反应的产物为甲醛、苯酚、2-羟基苯基乙醛和4-羟甲基苯酚等,主要为液体;后继反应中,热解产物与水蒸气发生反应,并得到CO、CO2和H2等小分子气体,以及苯酚和甲醇。
[1] 吴创之, 刘华财, 阴秀丽. 生物质气化技术发展分析[J]. 燃料化学学报, 2013, 41(7): 798-804. (WU Chuangzhi, LIU Huacai, YIN Xiuli. Status and prospects for biomass gasification[J].Journal of Fuel Chemistry and Technology, 2013, 41(7): 798-804.)
[2] CARLOS L. High-temperature air/steam gasification of biomass in an updraft fixed bed batch type gasifier[D].Stockholm: Royal Institute of Technology, 2005.
[3] YANG W, PONZIO A, LUCAS C, et al. Performance analysis of a fixed-bed biomass gasifier using high temperature air[J].Fuel Processing Technology, 2006, 87(3): 235-245.
[4] BOLTON R, PAN W. Lignin form rice straw Kraft pulping: Effects on soil aggregation and chemical properties[J]. Tetrahedron Lett, 2007, 98(7): 1482-1488.
[5] RAMONA D, YANG W, BLASIAK W. Wood pellets combustion with rich and diluted air in HTAC/G furnace[R].Stockholm: Report for EU-SUSPOWER Project, 2007.
[6] MAGNUS N, HAKAN E. Lignin: Recent advances and emerging applications[J].Current Opinion in Colloid & Interface Science, 2014, 19(5): 409-416.
[7] 路瑶, 魏贤勇, 宗志敏, 等. 木质素的结构研究与应用[J].化学进展, 2013, 25(5): 838-858. (LU Yao, WEI Xianyong, ZONG Zhimin, et al. Structural investigation and application of lignins[J].Progress in Chemistry, 2013, 25(5): 838-858.)
[8] CHEN Y R, SIMO S. Macromolecular replication during lignin biosynthesis[J].Phytochemistry, 2010, 71(4): 453-462.
[9] 谭洪, 王树荣, 骆仲泱, 等. 木质素快速热裂解试验研究[J].浙江大学学报(自然科学版), 2005, 39(5): 710-714. (TAN Hong, WANG Shurong, LUO Zhongyang, et al. Experimental study of lignin flash pyrolysis[J].Journal of Zhejiang University (Engineering Science), 2005, 39(5): 710-714.)
[10] 岳金方, 应浩. 工业木质素的热裂解试验研究[J].农业工程学报, 2006, 22(增刊1): 125-128. (YUE Jinfang, YING Hao. Experimental study on industrial lignin pyrolysis[J].Transactions of the CSAE, 2006, 22(Supp 1): 125-128.)
[11] QIN Y H, HUANG H F, WU Z B, et al. Characterization of tar from sawdust gasified in the pressurized fluidized bed[J].Biomass and Bioenergy, 2007, 35(4): 397-400.
[12] 刘江燕. 木质素及其模型物在不同热化学环境下的解构[D].广州: 华南理工大学, 2010.
[13] 王华静, 赵岩, 王晨, 等. 木质素二聚体模型物裂解历程的理论研究[J].化学学报, 2009, 67(9): 893-900. (WANG Huajing, ZHAO Yan, WANG Chen, et al. Theoretical study on the pyrolysis process of lignin dimer model compounds[J].Acta Chimica Sinica, 2009, 67(9): 893-900.)
[14] ELDER T, BEATE A. Density functional theory study of the concerted pyrolysis mechanism for lignin models[J].Energy & Fuels, 2014, 28(8): 5229-5235.
[15] 黄金保, 武书彬, 雷鸣, 等. 木质素二聚体模型化合物热解机理的量子化学研究[J].燃料化学学报, 2015, 43(11): 1334-1343. (HUANG Jinbao, WU Shubin, LEI Ming, et al. Quantum chemistry study on pyrolysis mechanism of lignin dimer model compound[J].Journal of Fuel Chemistry and Technology, 2015, 43(11): 1334-1343.)
[16] HUANG J B, HE C, LIU C, et al. A computational study on thermal decomposition mechanism ofβ-1 linkage lignin dimer[J].Computational and Theoretical Chemistry, 2015, 1054(2): 80-87.
[17] 武书彬, 刘超, 邓裕斌.α-O-4型木质素二聚体热解行为的理论研究[J].华南理工大学学报(自然科学版), 2015, 43(6): 22-29. (WU Shubin, LIU Chao, DENG Yubin. Theoretical investigation into pyrolysis behaviors ofα-O-4 lignin dimer[J].Journal of South China University of Technology(Natural Science Edition), 2015, 43(6): 22-29.)
[18] 曹小玲, 张航, 邓胜祥, 等.β-1型木质素二聚体高温蒸汽气化机理的理论研究[J].燃料化学学报, 2014, 42(7): 813-819. (CAO Xiaoling, ZHANG Hang, DENG Shengxiang, et al. Theoretical study on gasification ofβ-1 type lignin dimer with high temperature steam[J].Journal of Fuel Chemistry and Technology, 2014, 42(7): 813-819.)
[19] 王继仁, 孙艳秋, 邓存宝, 等. 煤自燃生成水的反应机理研究[J].煤炭转化, 2008, 31(1): 51-56. (WANG Jiren, SUN Yanqiu, DENG Cunbao, et al. Coal spontaneous combustion producing water reaction mechanism research[J].Coal Conversion, 2008, 31(1): 51-56.)
[20] 叶蔚甄, 龙军, 赵毅, 等. 环烷酸催化脱羧机理的研究[J].石油学报(石油加工), 2016, 32(5): 943-950. (YE Weizhen, LONG Jun, ZHAO Yi, et al. Study on the mechanism of catalytic decarboxylation of naphthenic acid[J].Acta Petrolei Sinica(Petroleum Processing Section), 2016, 32(5): 943-950.)
[21] BICOUT D, FIELD M. Quantum mechanical simulation method for studying biological system[M].Berlin: Les Houches Workshop, 1995: 1-21.
[22] KAREN L S, BRETT T D, TODD E, et al. Basis set exchange: A community database for computational sciences[J].Chem Inf Model, 2007, 47(3): 1045-1052.
[23] MOSTAGHNI F, TEIMOURI A, MIRSHOKRAEI S A. Synthesis, spectroscopic characterization and DFT calculation ofb-O-4 type lignin model compounds[J].Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 2013, 110(6): 430-436.
[24]黄金保. 纤维素快速热解机理的分子模拟研究[D].重庆: 重庆大学, 2010.
[25] TAMMARAT P, NAWEE K, SIRIPOM J. Application of the reaction class transition state theory to the kinetics of hydrogen abstraction reactions of alkanes by atomic chlorine[J].Computational and Theoretical Chemistry, 2013, 1011(5): 65-74.
[26] TOKIZAKI C, YOSHIDA T, TAKAYANAGI T. Quantum transition state dynamics of the cyclooctatetraene unimolecular reaction on ab initio potential energy surfaces[J].Chemistry Physics, 2016, 469-470(5): 97-104.
DensityFunctionalTheoryofGasificationReactionofLignanModelCompoundWithβ-5Linkage
ZHANG Hang1, DENG Shengxiang1, TIAN Hong2, CAO Xiaoling2
(1.SchoolofEnergyScienceandEngineering,CentralSouthUniversity,Changsha410083,China;2.SchoolofEnergyandPowerEngineering,ChangshaUniversityofScienceandTechnology,Changsha410015,China)
2016-10-10
国家自然科学基金项目(51276023)和国家自然科学基金国际(地区)合作与交流项目(513101410)资助
张航,男,博士研究生,从事煤和生物质热解气化方面的研究;E-mail:zhanghang4202@126.com
1001-8719(2017)05-0975-10
TK 224
A
10.3969/j.issn.1001-8719.2017.05.021
