基于硫化物固态电解质的固态锂硫电池研究进展
2017-10-14孙滢智黄佳琦张学强
孙滢智,黄佳琦,张学强,张 强
基于硫化物固态电解质的固态锂硫电池研究进展
孙滢智1, 2,黄佳琦1, 3,张学强1,张 强1
(1清华大学化学工程系,绿色反应工程与工艺北京市重点实验室,北京 100084;2清华大学材料学院,北京 100084;3北京理工大学前沿交叉科学研究院,材料学院,北京 100081)
便携式电子设备、电动汽车、智能输电网络等的发展对电化学储能系统提出了新的要求和挑战。固态锂硫电池因其高安全性和极高理论能量密度而受到广泛关注。硫化物固态电解质拥有高离子导率和优异的界面接触特性,适用于构建高性能的固态锂硫电池。本文从硫化物固态电解质的性质出发,针对正极侧高效载流子通道构筑和负极侧金属锂/电解质界面稳定性等问题,系统地阐述了基于硫化物固态电解质的固态锂硫电池研究进展,并指出未来的研究关键和重要发展方向。
锂硫电池;固态电解质;离子导率;硫化物;金属锂
随着便携式电子设备的普及、电动汽车及混合动力汽车等的应用和推广,当今社会对于电化学储能器件的需求与日俱增,并提出了新的挑战[1]。传统的锂离子电池受制于电极材料较低的理论容量,能量密度进一步提升的空间有限,难以支撑下一代电子设备对高能量密度储能系统的要求。因此,寻找高性能的、新体系的电池系统变得尤为重要[2]。
锂硫电池正极活性物质硫具有很高的理论比容量(1672 mA·h/g),此外硫还具有矿藏丰富、本身毒性小、环境友好的优势。金属锂拥有最低的标准电极电势(-3.04 V. 标准氢电极)和很高的理论容量密度(3860 mA·h/g),是理想的负极材料。因此,锂硫电池有望作为新一代的高能量密度电池系统得到广泛应用。
随着电池储能规模的逐渐扩大、应用日益广泛,电池的安全性也变得越发重要。传统的电池体系大多应用液态电解液,存在着电解液渗漏、燃烧、爆炸等危险[2-3]。为了解决此类问题,近年来新型固态电解质的开发以及固态电池的设计正逐渐成为研究的热点,许多研究组,如中国科学院宁波材料技术与工程研究所的许晓雄研究员及其团队等[4-5],在该领域做出了十分优秀的工作,进一步促进了固态电池的发展。
其中对于锂硫电池体系,固态电解质相比于液态电解液还存在着3个明显的优势:①应用固态电解质可以避免放电中间产物多硫化物的溶解和飞梭效应的产生;②在固态电解质中只有Li+可以迁移,即迁移数为1,有利于调控负极侧金属锂的均匀沉积、抑制锂枝晶生长;③ Li+在固态电解质/电极材料界面上的传递和转移不涉及去溶剂化的过程,相应的活化势垒较低,有利于电荷在界面处的快速传输。
对于固态电解质而言,高离子导率是需要满足的首要条件。目前为止,人们已研发出了多种在室温下离子导率高于10-4S/cm的固态电解质材料,其中硫化物类固态电解质和氧化物类固态电解质应用最为广泛。如石榴石结构的Li7La3Zr2O12[6]、NASICON结构的Li1.3Al0.3Ti1.7(PO4)3[7]等氧化物类固态电解质拥有着较高的室温离子导率(10-3~10-4S/cm),可以在空气中直接制备和操作。但是由于氧化物固态电解质本身硬度较大、界面接触较差,所以简单的冷压难以有效地减小晶界阻力和电解 质/活性材料界面的阻力,而必须通过高温烧结才能实现较高的离子导率。但是在烧结温度下活性物质、固态电解质以及导电剂间会发生反应,所以,对于锂硫电池而言,氧化物固态电解质的实际应用性并不强。相比之下,硫化物固态电解质硬度较低,界面接触较好,制得的粉末仅需冷压便可有效地降低界面阻抗、获得较高的离子导率,这也使硫化物固态电解质在固态锂硫电池中得到了广泛的应用[8]。本文将评述基于硫化物固态电解质的固态锂硫电池研究状况。
1 硫化物固态电解质
由于S2-的离子半径比O2-大很多,所以S2-对氧化物固态电解质结构中的O2-进行取代时,可以有效地扩大Li+的传输通道。同时由于S2-相对于O2-更易极化,相应的阴离子骨架与Li+之间作用力较弱,有利于Li+的迁移。这些优势使得许多硫化物电解质在室温下都具有很高的Li+导率(表1)[7-26]。根据结构及性质的不同,本文把在固态锂硫电池中有所应用的硫化物固态电解质大致分为Li2S-P2S5玻璃陶瓷、thio-LISICON、阴离子掺杂Li2S-P2S5等。
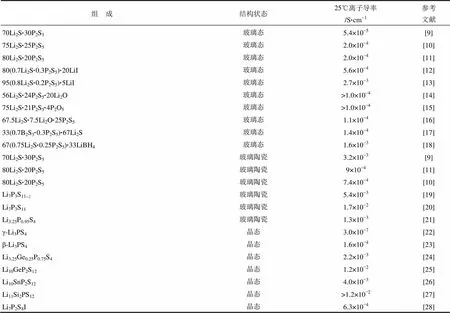
表1 不同硫化物固态电解质的离子导率
1.1 Li2S-P2S5玻璃及玻璃陶瓷
Li2S-P2S5玻璃是一类研究十分广泛的固态电解质,许多研究致力于提升该类材料的离子导率,其中通过退火得到玻璃陶瓷(微晶玻璃)是一种有效的提高其离子导率的方法。HAYASHI等[11]通过对S和Li2S进行高能球磨获得了室温离子导率为2×10-4S/cm的玻璃相80Li2S·20P2S5,进一步在250 ℃下加热退火后80Li2S-20P2S5的离子导率提高至了9×10-4S/cm,离子导率的提高是由于退火过程中80Li2S-20P2S5的结晶度提高。
进一步的研究表明,Li2S-P2S5玻璃陶瓷的离子导率高低与退火温度有很大关系,除了结晶程度的影响外,在不同温度下退火所得到的Li2S-P2S5玻璃陶瓷组成实际上也是不同的。图1为80Li2S-20P2S5玻璃在不同温度下退火后的X射线衍射(XRD)图谱。从中可以看出,在240 ℃退火后可以获得离子导率很高的thio-LISICON相,如Li3.25P0.95S4,而当退火温度升高至500 ℃后,80Li2S-20P2S5玻璃陶瓷中主要成分则转变为热力学稳定但离子导率相对较低的Li7PS6和Li3PS4[10]。MIZUNO等[9]发现球磨制得的70Li2S-30P2S5玻璃在240 ℃下热处理2 h后有新相生成,由此获得的70Li2S-30P2S5玻璃陶瓷的离子导率高达3.2×10-3S/cm。TATSUMISAGO等[29]进一步证明70Li2S-30P2S5玻璃在240 ℃下热处理得到的新相为Li7P3S11,加热至360 ℃可提高Li7P3S11结晶度,但若继续加热至550 ℃以上,Li7P3S11相便会基本消失并转化为热力学稳定的Li4P2S6。由于Li4P2S6的离子导率很低(约10-6S/cm),所以550 ℃退火后70Li2S-30P2S5玻璃的离子导率明显下降。
除了电解质材料的内在性质,界面条件对于获得高的离子传输率也十分重要。SEINO等[20]将70Li2S·30P2S5玻璃在94 MPa的高压下压实后在280 ℃下热处理2 h,有效地消除了离子迁移的晶界阻力。通过此方法制得的70Li2S-30P2S5玻璃陶瓷的室温离子导率可以达到1.7×10-2S/cm,超过常用液态电解液的离子导率。从扫描电子显微镜(SEM)照片可以看出,仅通过简单冷压得到的70Li2S-30P2S5玻璃陶瓷内部实际上还存在着许多间隙和晶界,而经过热处理后此类间隙和晶界明显变少,阻抗也相应地大幅降低(图2)。
1.2 Thio-LISICON类固溶体
Thio-LISICON是最为典型的一类晶态硫化物固态电解质,化学通式可以表示为Li4-A1-BS4(A=Si、Ge等,B=Zn、Al、P等)。KANNO等[30]最早在Li2S-GeS2、Li2S-GeS2-ZnS和Li2S-GeS2- Ga2S3体系中制得了相应的thio-LISICON,并对比了不同组成thio-LISICON的离子导率。他们发现合理地用异价元素进行取代可以有效地提高thio-LISICON的离子导率。Li4GeS4在室温下的离子导率很低(2×10-7S/cm),但如果通过Ge4+Ga3++Li+的方式进行取代,所得到的Li4.275Ge0.61Ga0.25S4在室温下的离子导率可达6.5×10-5S/cm。在此基础上,KANNO等[24]进一步尝试通过Ge4++Li+P5+的方式进行取代得到thio-LISICON Li4-Ge1-PS4(0<<1.0)。通过XRD图谱可以看出,根据结构的不同thio-LISICON Li4-Ge1-PS4可以被分为3个组成部分:区域Ⅰ (0<≤0.6),区域Ⅱ6<≤0.8),区域Ⅲ(0.8<<1.0)(图3)。其中区域Ⅱ(0.6<≤0.8)所对应的thio- ISICON相拥有特殊的单斜超晶格结构以及更高的室温离子导率(>10-3S/cm)。他们认为相比于Li4GeS4,thio-LISICON Li4-Ge1-PS4更高的离子导率归功于异价元素取代后所形成的Li+空穴。
2011年,KANNO等[31]成功合成了一种室温离子导率(1.2×10-2S/cm)与液态电解液相当的硫化物固态电解质Li10GeP2S12。图4(a)和图4(b)为Li10GeP2S12的晶体结构,其整体上是一个由(Ge0.5P0.5)S4四面体、PS4四面体、LiS4四面体以及LiS6八面体构成的三维离子通道。Li10GeP2S12晶胞中有4和2两种四面体位置,其中4四面体位置被Ge和P占据,而较小的2四面体位置则只被P所占据。Li则占据16、4和8三个位置。(Ge0.5P0.5)S4四面体和LiS6八面体通过共边的方式沿轴构建一维长链,各个一维长链之间通过PS4四面体相互连接组成三维骨架,而其中一维的离子通道则由位于16和8的LiS4共边连接而成[25]。WANG等通过计算推测Li10GeP2S12中存在体心立方阴离子亚晶格[图4(c)]。在体心立方亚晶格中,Li+从一个四面体位置迁移至另一个共面的四面体位置,这种迁移路径的能垒很低,有利于Li+在Li10GeP2S12中的快速传输[图4(d)]。
尽管Li2S-GeS2-P2S5体系的thio-LISICON已经能够拥有很高的室温离子导率,但是由于其与金属锂之间较差的化学匹配性和锗昂贵的价格[32],寻找其它硫化物,如SnS2、SiS2,来代替GeS2合成具有高离子导率的thio-LISICON仍十分重要。对于Li2S-SnS2-P2S5体系,Sn的掺杂量十分有限,最多也只能得到Li10SnP2S12[26]。而对于Li2S-SiS2-P2S5体系,由于更多的P5+可以被替换为Si4+,因此掺杂量可以更多,最多可以得到Li11Si2PS12。KUHN等[33]认为这是由于Si4+的离子半径小,可以占据较小的2四面体位置。同时,Si4+替换P5+后为了补偿损失的正电荷,晶体内部Li+的含量将增加,这将有利于获得高的离子导率[27]。ONG等[31]也通过计算证明了将Li10GeP2S12中的Ge替换为Si将让离子迁移活化能降低0.01 eV,而用Sn替换Ge将让离子迁移活化能升高0.03 eV。
1.3 阴离子掺杂Li2S-P2S5
通过掺杂另一种阴离子的方法可以有效地提高硫化物玻璃的离子导率,这也被称为“混合阴离子效应”。早在1981年,MERCIER等[34]便通过在Li2S-P2S5中掺杂LiX(X=Cl, Br, I)的方法获得了室温离子导率达到10-4S/cm的固态电解质。同时他们还发现了离子导率与掺杂阴离子的极化之间的正相关性(LiI>LiBr>LiCl)。UJIIE等[12-13]进一步合成并研究了(100-)(0.7Li2S·0.3P2S5)·LiI、(100-) (0.8Li2S·0.2P2S5)·LiI玻璃及玻璃陶瓷电解质。他们发现对于(100-)(0.7Li2S·0.3P2S5)·LiI玻璃,离子导率随LiI含量增加而升高;而对于(100-) (0.7Li2S·0.3P2S5)·LiI玻璃陶瓷,掺入LiI将使得 离子导率明显降低。相比之下,(100-) (0.8Li2S·0.2P2S5)·LiI玻璃陶瓷的离子导率则随LiI的增加先升高后降低,但整体上保持在一个很高的水平,当=5时离子导率最高(2.7×10-3S/cm)。
此外,阴离子掺杂也可以提高硫化物固态电解质的稳定性。EZHIYLMURUGAN等[28]通过对β-Li3PS4和LiI进行混合得到了新的Li7P2S8I相。虽然Li7P2S8I室温下的离子导率并不是很高(6.3×10-4S/cm),但其电化学稳定窗口可以高达10 V(. Li/Li+)[图5(a)]。他们认为新相中I-很好地融合进了整体的固溶结构当中,从而弥补了I-易被氧化的缺陷。同时,I的存在又提高了硫化物电解质与金属锂之间的稳定性,Li/Li7P2S8I/Li对称电池在恒流充放电的过程中十分稳定[图5(b)],这对于其在金属锂电池中的应用有重要意义。
(a)
(b)
图5 (a)Li/ Li7P2S8I/Pt电池在1 mV/s扫描速度下的循环伏安曲线;(b)Li/ Li7P2S8I/Li对称电池在0.2 mA/cm2电流密度下的直流极化曲线[28]
Fig.5 (a) Cyclic voltammogram for a Li/ Li7P2S8I/Pt cell at a scan rate of 1 mV·s−1; (b) DC polarization curve for a Li/ Li7P2S8I/Li symmetric cell at a current density of 0.2 mA·cm−2[28]
大多数硫化物电解质对水都不稳定而且十分敏感,这也导致了其实际的制备和应用不能简单地在空气中进行。为了克服这一缺陷,很多研究尝试通过O掺杂来部分地取代S进而提高硫化物电解质对于水的稳定性。如OHTOMO等[14]报道用Li2O部分地取代70Li2S·30P2S5中的Li2S能有效地抑制水解及H2S的产生;HAYASHI等[15]也曾运用P2O5取代P2S5来抑制硫化物玻璃电解质暴露在空气中后与水反应的速率。然而,此类掺杂都会在体系中引入PO4单元和硫氧单元,其中的非桥接氧对Li+有很大的束缚作用,因此会明显降低固态电解质的离子导率。如何在保证高离子导率的情况下提高硫化物电解质对水蒸气的稳定性仍有待进一步的研究。
2 正极侧结合硫化物固态电解质的 研究及应用
虽然此前的研究已经制备出了多种高离子导率的硫化物固态电解质,其中一些的导离子性甚至和液态电解质相当,不过要将硫化物固态电解质应用到锂硫电池当中还需考虑锂硫电池本身的一些特性。
单质硫作为活性物质虽然拥有很高的理论容量,但是作为电极材料单质硫却存在着3个主要的弊端:①活性物质S及放电产物Li2S的导电性和导离子性很差;②放电过程中,从S转化为Li2S存在着很大的体积膨胀(约79%),破坏电极结构;③对于常用的液态电解液体系,充放电过程中产生的高阶多硫化物会溶解到电解液当中,进而扩散至负极与金属锂直接反应变为低阶多硫化物又扩散回正极与单质硫生成高阶多硫化物,如此往复循环便形成了所谓的“飞梭效应”,这将严重损害电池的容量及库仑效率。
尽管使用固态电解质可以从根本上避免“飞梭效应”的发生,但因为固态电解质本身不具备流动性,所以充放电过程中的大幅体积变化不仅会破坏电极中的电子通道,还会破坏离子通道。同时可溶性氧化还原中间体(多硫化物)的缺失也使得活性物质S及放电产物Li2S在导电性和导离子性方面的劣势变得更为突出。因此,构筑高效的离子-电子通道,改善活性物质与电解质,导电骨架间离子及电子的转移对于有效利用活性物质和提升固态锂硫电池性能尤为重要。为了在电极内部构筑快速、有效的载流子传输通道,此前的研究已经尝试了多种硫化物电解质及导电剂体系来搭建固态锂硫电池的正极(表2),在容量密度和循环性能上都取得了很大的进展。活性物质与固态电解质间离子的有效传输作为其中的关键步骤而得到了广泛的研究和关注,本文将就此方面工作进行重点介绍。
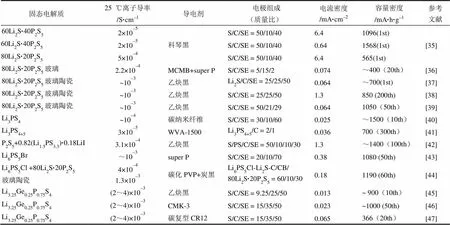
表2 应用不同硫化物固态电解质的锂硫电池性能
2.1 纳米化促进离子传输
提升活性物质与电解质之间离子迁移的一个重要方法是将活性物质纳米化或附着在纳米结构骨架上,利用纳米材料的高比表面积使活性物质与固态电解质充分接触,进而整体上增强活性物质与固态电解质间的离子迁移。同时,由于S和Li2S本身离子导率和电子导率很低,在没有可溶性多硫化物产生的情况下电化学反应能够进行的深度有限,此时利用纳米化的手段可以很大程度地提高活性物质的利用效率。
NAGAO等[37]通过预先对Li2S进行单独球磨,再将得到的Li2S与Li2S-P2S5玻璃陶瓷、乙炔黑进一步球磨来制备纳米复合正极。通过此方法制得的纳米复合正极中活性物质Li2S的颗粒尺寸在500 nm左右,并且能够很均匀地分散在导电剂和固态电解质当中。在充放电循环后,Li2S颗粒的尺寸依然可以保持纳米尺度而不发生团聚[图6(a)~(d)]。在0.064 mA/cm2(0.035 C)的电流密度下,纳米Li2S复合正极的循环可逆容量可以达到1000 mA·h/g。通过与未经过预球磨的Li2S普通正极材料进行对比,可以发现减小活性物质颗粒尺寸对于提高电池容量有显著作用,并且随着电流密度的升高这种优势会越发明显,在6.4 mA/cm2(3.5 C)的高倍率条件下,纳米复合正极依然拥有271 mA·h/g的容量,而普通正极则仅有70 mA·h/g左右的容量。为了使Li2S、乙炔黑以及固态电解质获得更好的混合与接触,NAGAO等[39]进一步使用在155 ℃加热球磨的方法来制备纳米复合正极。通过此种方法制备的纳米复合正极中活性物质S颗粒的尺寸更小(<200 nm),相应的与导电剂、固态电解质之间有更充分的接触[图6(e)~(f)]。在此基础上可以制备出硫含量高达50%的复合正极材料,该硫含量基本是此前固态锂硫电池正极硫含量的2倍。同时,此高硫含量正极依然有着十分不错的可逆容量和循环稳定性,在0.064 mA/cm2的电流密度下循环50圈后依然可以拥有1050 mA·h/g的容量以及1007 W·h/kg的质量能量密度。相比之下,通过常温球磨获得的复合正极在如此高的硫含量条件下首圈容量仅有550 mA·h/g左右,远低于经过加热球磨得到的复合正极。这与加热球磨过程中获得了尺寸更小的纳米Li2S颗粒以及其与固态电解质及导电剂更好的接触密切相关。
由于固态电解质不具备流动性和渗透性,而Li2S和S的导离子性又很差,所以在固态锂硫电池正极中离子通道和电子通道很大程度上是隔离的,即活性物质得失电子的部位不能即时地导入或导出离子,这可能造成局部电荷堆积而阻碍电化学反应的进行。纳米化之后的活性物质因为可以在一个很小的尺寸范围内同时接触到导电剂和固态电解质,所以能够一定程度打破这种隔阂实现电子-离子混合导通。HAN等[44]报道了一种自下而上的方法来合成活性物质、固态电解质以及导电骨架的纳米复合物从而实现了电子和离子的混合导通。他们将聚乙烯吡咯烷酮、Li6PS5Cl以及Li2S溶解到乙醇中,进一步通过共沉淀和高温碳化的过程使得活性物质Li2S和固态电解质Li6PS5Cl以纳米颗粒的形式均匀分散到碳骨架当中[图7(a)]。通过高分辨投射电镜可以看出其中Li2S颗粒和Li6PS5Cl颗粒尺寸在4 nm左右,高倍率照片显示Li2S颗粒的确可以在一个很小的尺寸内同时与C和Li6PS5Cl相互接触从而达到电子-离子混合导通的效果[图7(b)]。将Li2S- Li6PS5Cl-C纳米复合物和Li2S-C纳米复合物与炭黑及80Li2S-20P2S5混合制得相应正极材料应用到固态锂硫电池中,对比二者的热力学平衡电势可发现Li2S-Li6PS5Cl-C电极拥有更低的过电势[图7(c)],证明了电子-离子混合通道的存在有助于提高充放电过程中电极反应的动力学。另一方面电子-离子混合通道的存在也提高了活性物质的利用率,在50 mA/g的电流密度下,Li2S-C电极的初始可逆容量仅有489 mA·h/g(此工作中容量均基于Li2S质量计算)并且循环至第10圈开始快速衰减,Li2S-C电极第20圈的容量仅剩49 mA·h/g。相比之下,Li2S- Li6PS5Cl-C电极首圈容量便可以达到648 mA·h/g,循环60圈后容量保持在830 mA·h/g[图7(d)][44]。
除了对活性物质进行直接的纳米化,利用一些纳米结构作为骨架来负载活性物质也是一种有效的提升活性物质与固态电解质及导电剂接触的方式。纳米骨架对于活性物质在放电过程中的膨胀也有一定抑制作用,有利于保持固态电解质与正极活性物质在循环过程中持续的有效接触。KANNO课题组[46]以介孔氧化铝为模板制备了孔径为1~2 nm的介孔碳材料CMK-3。通过在300 ℃下气相混合的方法将S负载至CMK-3的纳米孔道当中,冷却后再升温至230 ℃来除去孔道结构外的S得到S/CMK-3复合物,进一步将S/CMK-3复合物与thio-LISICON(Li3.25Ge0.25P0.75S4)球磨混合作为正极材料。通过此种方法制得的正极材料拥有很好的循环性能和容量密度,在0.023 mA/cm2(0.09 C)的电流密度下,循环20圈和50圈后容量仍能达到1300 mA·h/g和1000 mA·h/g。相比之下,用未除去纳米孔道外S的S/CMK-3复合物制得的正极材料在同等电流密度下循环20圈后容量仅有500 mA·h/g左右,由此可以看出利用纳米骨架负载活性物质可以有效地提高活性物质的利用率。虽然运用CMK-3的介孔结构说明了纳米负载骨架对于提升固态锂硫电池性能的积极作用,然而由于CMK-3的二维排列碳棒结构仅能提供单一尺寸的纳米孔道,所以该工作对于何种结构及尺寸的纳米骨架更有利于电极与固态电解质之间的接触并没有进行深入的探讨。针对这一问题,KANNO课题组[47]进一步研究了纳米骨架中孔道尺寸对于固态锂硫电池正极性能的影响。他们利用不同尺寸(12 nm、40 nm和100 nm)的纳米二氧化硅球作为模板制备了拥有相应尺寸三维孔道结构的碳复型(分别以CR12、CR40、CR100表示),进一步利用气相混合的方法制得相应的S/CR复合物[图8(a)],S/CR复合物和Li3.25Ge0.25P0.75S4通过球磨混合制得正极材料。在0.064 mA/cm2的电流密度下,经过20圈恒流充放电循环后,S/CR12、S/CR40和S/CR100分别保有366 mA·h/g、126 mA·h/g和57 mA·h/g的容量密度[图8(b)]。在1.3 mA/cm2的大电流密度下,S/CR12仍有489 mA·h/g的初始容量,而S/CR40和S/CR100的初始容量均不到180 mA·h/g[图8(c)]。由此可以看出,在100~10 nm的孔径范围内,纳米孔道尺寸越小,容量密度和倍率性能越好,这得益于小尺寸时活性物质与固态电解质更有效的接触。
2.2 通过改善界面促进离子传输
活性物质的纳米化主要是依靠增加活性物质与固态电解质接触面积的方式,在整体上提升活性物质和固态电解质间的离子迁移,而并没有本质上改变离子在二者间迁移的速率。相比之下,另一种提升活性物质和固态电解质间离子迁移的方法则是通过改善活性物质/固态电解质界面来加速离子迁移的速率。
HIROSHI等[35]研究了应用不同固态电解质的锂硫电池容量,从中他们发现锂硫电池的容量并非与固态电解质的离子导率完全呈正相关。尽管Li1.5PS3.3(60Li2S-40P2S5)的离子导率(2×10-5S/cm)远低于Li4.0PS4.5(80Li2S-20P2S5)(5×10-4S/cm),然而在6.4 mA/cm2的电流密度下, Li1.5PS3.3作为电解质的锂硫电池容量可达1096 mA·h/g,而Li4.0PS4.5作为电解质的锂硫电池容量仅有565 mA·h/g。他们认为这是由于Li1.5PS3.3对于S起到了活化的作用使其反应性提高。在此基础上,HIROSHI等[42]进一步将0.82(Li1.5PS3.3)·0.18(LiI)和P2S5两种硫化物结合用以构建复合正极。其中0.82 (Li1.5PS3.3)·0.18(LiI)拥有着很高的离子导率(3.1×10-3S/cm)用以提供离子通道,P2S5则用来提高S的反应活性。由此制得的复合正极在1.3 mA/cm2的电流密度下初始容量高达1550 mA·h/g,即使在100个循环后容量仍能达到约1400 mA·h/g。虽然取得了很好的电池性能,但HIROSHI等所提出的“活化”概念过于模糊,并没有解释清楚P2S5能活化S的本质原因。根据其后的相关研究,本文认为在充放电的过程中产生的Li2S可能原位与其表面的P2S5反应转化为了Li3PS4。Li3PS4本身拥有着很高的Li+导率,再加之是原位生成与活性物质颗粒本身有着很好的接触,因此大幅度提升了活性物质和固态电解质界面处离子迁移的效率,使得活性物质得到了更充分的利用,从而起到了所谓的“活化”作用。
在活性物质纳米化的基础上进一步加快界面Li+迁移速率将使固态锂硫电池的性能获得更大的提升。LIN等[48]通过将S与Li(CH2CH3)3BH在四氢呋喃中反应获得纳米Li2S颗粒,进而将得到的纳米Li2S与P2S5反应使纳米Li2S表面包覆一层Li3PS4,这层固态电解质外壳有效地提升了Li+向活性物质的迁移速率,使得纳米Li2S颗粒在室温下的离子导率提高了4个数量级。应用Li3PS4包覆的纳米Li2S颗粒(以下简称“LSS颗粒”)作为正极材料,在60 ℃、0.1 C下首圈放电容量能达到848 mA·h/g(此工作中容量均基于Li2S质量计算),循环100圈后容量仍能保持在594 mA·h/g,相同条件下只运用纳米Li2S颗粒作为正极材料首圈放电容量仅有569 mA·h/g,循环100圈后容量仅剩402 mA·h/g[图9(a)]。同时,对比二者的充放电曲线也可以看出运用LSS颗粒作为正极材料时的过电势明显减小[图9(b)],这都得益于包覆Li3PS4后界面处离子的迁移效率得到了提升。
2.3 提高活性物质本身离子传输能力
除了改善活性物质和固态电解质间界面的离子迁移,改变活性物质S和Li2S本身很差的离子导率、保证Li+在活性物质内部的快速传输同样十分重要。LIN等[41]通过将单质硫与Li3PS4在四氢呋喃(THF)中混合反应,在中引入多硫链从而得到了多硫代磷酸盐Li3PS4+n(0<<9)。由此制得的多硫代磷酸锂拥有好的导离子性,Li3PS4+5在25 ℃的离子导率可以达到3×10-5S/cm,远高于S和Li2S(约10-13S/cm)。由于其出色的导离子性,Li3PS4+5在正极中可以兼顾活性物质和固态电解质两方面的作用。应用Li3PS4+5作为正极材料的固态锂硫电池拥有着很高的容量和循环稳定性能,室温下,在0.1 C测试条件下初始容量可以达到1272 mA·h/g(本段中容量均是基于未与P键合的S质量),循环300圈后容量保持在700 mA·h/g。在60 ℃下该电池的循环性能更为突出,初始容量高达1400 mA·h/g,循环300圈后容量仍可以保持在1200 mA·h/g。同时,通过对比初始状态、放电及充电后正极材料的拉曼光谱可以发现其中的确存在着S—S键的断裂和再形成,这一定程度上证明了Li3PS4+n与Li2S、Li3PS4间的转化在循环过程中是可逆的。该工作利用硫化物电解质与单质硫间的化学键合为固态锂硫电池正极的构建提供了新的思路,同时也证明了提升活性物质本身离子导率的重要作用。
3 负极侧结合硫化物固态电解质的 研究和应用
金属锂负极是锂硫电池发展和应用的另一大挑战,有关金属锂负极的研究可以追溯到20世纪70年代[49]。金属锂由于本身拥有高达3860 mA·h/g的理论比容量和最低的氧化还原电势(约3.04 V标准氢电极)而被认为是高能电池系统最理想的负极材料之一。然而金属锂负极在实际应用中却存在着许多严重的问题。锂的沉积和脱去过程并不是均匀的,这将导致锂枝晶的产生,锂枝晶的生长有可能刺穿隔膜导致电池短路等安全问题。同时,目前锂硫电池常用的电解液会与金属锂发生反应而损耗,这一方面导致活性金属锂材料循环效率较差,也使得在实用化的锂硫电池中必需添加足够的电解液以维持电池的正常工作,进一步导致了电池整体容量密度的降低[3, 50-51]。对于固态锂硫电池,金属锂的这些问题依然是需要考虑的,因此负极侧结合硫化物固态电解质的研究主要集中在金属锂和电解质的稳定性、锂离子在电解质/负极界面处的迁移以及金属锂的均匀沉积和脱去。值得注意的是,这3方面因素是相互关联和影响的,它们共同决定了固态电解质与金属锂的匹配性。
有关硫化物固态电解质和金属锂之间的稳定性,大阪府立大学的TATSUMISAGO课题组的研究表明,静置的Li/80Li2S·20P2S5/Li对称电池的阻抗随时间先升高后维持基本不变。YAMADA等[40, 52]也发现Li3PS4存在着类似的现象。他们认为这是因为硫化物固态电解质表面与金属锂反应生成了一层中间相,这层稳定的中间相阻止了金属锂与固态电解质的进一步反应。
然而对于实际的充放电过程,这种类型的稳定并不一定适用,还需考虑金属锂的沉积状态以及这层中间相的致密程度等因素的影响。如果锂沉积并不均匀进而形成锂枝晶,同时中间相又较为疏松,那么这层中间相很容易被锂枝晶刺破而毁坏,将不再能起到稳定固态电解质与金属锂界面的作用。通过改善金属锂与固态电解质间接触可以促进锂的均匀沉积进而保证二者界面的稳定,NAGAO等[53]通过真空蒸镀的方法在80Li2S-20P2S5电解质层两侧表面上覆盖了一层很薄的锂膜,使用这种经过修饰的电解质层的Li-Li对称电池在恒流充放电循环中很稳定,电压无明显变化。相比之下,应用未经修饰的电解质层的Li-Li对称电池在恒流充放电循环中电压则一直降低难以稳定。本课题组[54]研究发现硫化的固态电解质中间相(SEI)相比于普通的SEI离子导率明显提高,SEI内部存在分布更为均匀、致密的离子传输通道,有利于金属锂的均匀沉积和脱去[图12(d)~(e)],进一步提高了金属锂的循环性能。应用硫化SEI的Li/Cu纽扣电池在1.0 mA/cm2的电流密度下循环200圈后库仑效率仍能保持在98%,而应用普通SEI的电池库仑效率已经衰减到了70%以下[图12(f)]。这种循环稳定性方面的优势在软包电池中体现的更为明显,对于锂金属负极的实际应用有很大帮助[图12(g)]。从中我们可以进一步看出:如果适当地对硫化物固态电解质和金属锂负极间界面的组成进行调控,还是可能获得一个有利于金属锂均匀沉积-脱去的稳定界面的。
(a)
(b)
图11 (a)室温下Li/80Li2S·20P2S5/Li对称电池的电化学阻抗谱(EIS)随时间变化情况[52];(b)在80 ℃温度下,Li/Li3PS4/Li对称电池的阻抗7天内的变化[40]
Fig.11 (a) Time dependence of complex impedance plots at room temperature for the symmetrical cell Li/80Li2S·20P2S5/Li; (b)cell impedance evolution of the Li/Li3PS4/Li symmetrical cell at 80 ℃ for seven days[40]
YAMADA等[40]对使用液态电解质和Li3PS4固态电解质的Li/Li对电池沉积-脱去锂前后的电化学阻抗谱进行了对比,从中他们发现应用液态电池的电荷传输阻力(CT)在沉积-脱去锂前后变化不大,而固态电池的电荷传输阻力相比于初始状态则明显减小。进一步他们计算了不同温度下两种电池沉 积-脱去锂前后的交换电流密度0,并在此基础上得到了两种电池沉积-脱去锂前后离子迁移的活化能。液态电池经历了沉积-脱去锂后离子迁移活化能没有明显变化(由68.4 kJ/mol降至67.6 kJ/mol),而固态电池在沉积-脱去锂后Li+活化能则明显降低(由52.1 kJ/mol降至44.5 kJ/mol),YAMADA等对此给出的解释是:在液态电解质中,沉积-脱去锂的过程中会在原始SEI基础上再形成一层有机的SEI,而在固态电解质中,沉积-脱去锂的过程破坏了原始SEI后金属锂与固态电解质直接接触,因此离子迁移的活化能明显降低。虽然此工作中YAMADA等未将最初金属锂与固态电解质间相互反应产生的稳定中间相考虑到原始SEI中,不过从中可以看出不加调控的沉积-脱去锂过程对于固态电解质/金属锂中间层的破坏是的确存在的。虽然在充放电循环的初期这种破坏似乎有助于提高Li+的迁移,但是一个不稳定的界面在循环过程中势必会造成金属锂的不断损耗和不均匀沉积,如此恶性循环将严重影响负极的工作效率和循环性能。
4 总结与展望
考虑到多种电子设备对于高能量密度电化学储能系统的需求以及电池安全性等问题,固态锂硫电池体系正逐渐成为当今研究的热点之一。硫化物固态电解质拥有着很高的离子导率,同时本身硬度小、界面接触好,无需高温烧结即可用于制作电解质层和电极复合材料,有望在固态锂硫电池体系中得到应用。
除了硫化物固态电解质的相关性质,构建固态锂硫电池还需考虑锂硫电池本身的一些特性。针对正极侧,由于固态电解质本身不具备流动性和可溶性中间体缺失,活性物质S和Li2S本身导电性和导离子性差的弊端变得格外突出,在正极内部构建快速、高效的离子通道和电子通道尤为重要。为实现该目标,一方面可以通过活性物质的纳米化或借助纳米导电骨架来增大活性物质与固态电解质和导电剂的接触面积,进而整体上促进离子和电子的传递;另一方面可以利用原位反应包覆活性物质等手段来改善电解质/活性物质界面性质,提高离子的迁移速率。此外,通过将活性物质S与硫化物固态电解质进行化学键联获得新型的高离子导率活性物质也是从本质上解决该弊端的重要方法。针对负极侧,循环过程中金属锂与硫化物固态电解质界面的稳定性是需要考虑的关键问题,其不仅涉及金属锂和硫化物固态电解质间的化学稳定性,还和二者间的离子迁移以及金属锂的沉积-脱去性质有关。
尽管硫化物固态电解质在锂硫电池中的应用已经取得了较大的进展,但仍存在许多问题有待解决,其后续的研究仍有很大的发展空间。首先,在硫化物固态电解质方面,除了提高离子导率外,通过掺杂等方式增强其对于空气中水、氧等的稳定性对于其进一步的实际应用尤为重要;其次,正极侧离子的高效传输及离子通道结构的保持仍然是一个挑战,通过原位反应包覆等合适的方法对活性物质和固态电解质进行复合,进而提升离子在界面处的迁移速度、在体积变化过程中保证固态电解质与活性物质有效接触以及实现电子和离子的混合导通、同步传输将有助于进一步提高活性物质的利用效率;第三,针对负极侧,可以通过对固态电解质或金属锂表面嫁接稳定的SEI等手段改善界面离子迁移性质、促进金属锂的均匀沉积,从而达到稳定固态电解质/金属锂界面的作用。
考虑到固态电解质不具备流动性、界面接触差等缺陷,半固态锂硫电池也是一个值得研究的方向。通过运用多硫化物浆料作为正极材料或在固态电解质与电极间引入少量液态电解质润湿二者界面等手段,可以有效地解决上述问题。适当地搭配固、液两类电解质可以使得半固态锂硫电池在抑制“飞梭效应”和锂枝晶等问题的同时拥有一个很好的电极/电解质界面进而有效地提高电池性能。
随着新材料的引入以及人们对内在机理理解的加深,应用硫化物电解质的固态锂硫电池的性能将获得更大幅度的提升,相信在未来其将作为一种新型的高容量、高安全性电池体系发挥重要作用。
[1] DUNN B, KAMATH H, TARASCON J M. Electrical energy storage for the grid: A battery of choices[J]. Science, 2011, 334(6058): 928-935.
[2] GOODENOUGH J B, KIM Y. Challenges for rechargeable Li batteries[J]. Chemistry of Materials, 2010, 22(3): 587-603.
[3] XU K. Nonaqueous liquid electrolytes for lithium-based rechargeable batteries[J]. Chemical Reviews, 2004, 104(10): 4303-4417.
[4] 许晓雄, 邱志军, 官亦标, 等. 全固态锂电池技术的研究现状与展 望[J]. 储能科学与技术, 2013, 2(4): 331-341.
XU X X, QIU Z Z, GUAN Y B, et al. All-solid-state lithium-ion batteries:State-of-the-art development and perspective[J]. Energy Storage Science and Technology, 2013, 2(4): 331-341
[5] YAO X Y, LIU D, WANG C S, et al. High-energy all-solid-state lithium batteries with ultralong cycle life[J]. Nano Letters, 2016, 16(11): 7148-7154.
[6] MURUGAN R, THANGADURAI V, WEPPNER W. Fast lithium ion conduction in garnet-type Li7La3Zr2O12[J]. Angewandte Chemie-International Edition, 2007, 46(41): 7778-7781.
[7] AONO H, SUGIMOTO E, SADAOKA Y, et al. Ionic-conductivity of the lithium titanium phosphate [Li1+xAlTi2-(PO4)3], [Li1+xScTi2-(PO4)3], [Li1+xYTi2-(PO4)3], [Li1+xLaTi2-(PO4)3] systems[J]. Journal of the Electrochemical Society, 1989, 136(2): 590-591.
[8] SAKUDA A, HAYASHI A, TATSUMISAGO M. Sulfide solid electrolyte with favorable mechanical property for all-solid-state lithium battery[J]. Scientific Reports, 2013(3): doi:10.1038/ srep02261.
[9] MIZUNO F, HAYASHI A, TADANAGA K, et al. New, highly ion-conductive crystals precipitated from Li2S-P2S5glasses[J]. Advanced Materials, 2005, 17(7): 918-921.
[10] TATSUMISAGO M. Glassy materials based on Li2S for all-solid-state lithium secondary batteries[J]. Solid State Ionics, 2004, 175(1): 13-18.
[11] HAYASHI A, HAMA S, MORIMOTO H, et al. High lithium ion conductivity of glass-ceramics derived from mechanically milled glassy powders[J]. Chemistry Letters, 2001, 30(9): 872-873.
[12] UJIIE S, HAYASHI A, TATSUMISAGO M. Structure, ionic conductivity and electrochemical stability of Li2S-P2S5-LiI glass and glass-ceramic electrolytes[J]. Solid State Ionics, 2012, 211: 42-45.
[13] UJIIE S, HAYASHI A, TATSUMISAGO M. Preparation and ionic conductivity of (100−)(0.8Li2S·0.2P2S5)·LiI glass-ceramic electrolytes[J]. Journal of Solid State Electrochemistry, 2013, 17(3): 675-680.
[14] OHTOMO T, HAYASHI A, TATSUMISAGO M, et al. Characteristics of the Li2O-Li2S-P2S5glasses synthesized by the two-step mechanical milling[J]. Journal of Non-Crystalline Solids, 2013, 364: 57-61.
[15] HAYASHI A, MURAMATSU H, OHTOMO T, et al. Improved chemical stability and cyclability in Li2S-P2S5-P2O5-ZnO composite electrolytes for all-solid-state rechargeable lithium batteries[J]. Journal of Alloys and Compounds, 2014, 591: 247-250.
[16] TSUJIWAKI W, HIGUCHI E, CHIKU M, et al. Electrochemical analyzing method of the charge transfer reaction at the interface between sulfide-based solid electrolyte and positive electrode material with microelectrode[J]. ECS Transactions, 2014, 58(25): 77-84.
[17] ZHANG Z, KENNEDY J H. Synthesis and characterization of the B2S3-Li2S, the P2S5-Li2S and the B2S3-P2S5-Li2S glass systems[J]. Solid State Ionics, 1990, 38(3/4): 217-224.
[18] YAMAUCHI A, SAKUDA A, HAYASHI A, et al. Preparation and ionic conductivities of (100−)(0.75 Li2S·0.25 P2S5)·LiBH4glass electrolytes[J]. Journal of Power Sources, 2013, 244: 707-710.
[19] HAYASHI A, MINAMI K, UJIIE S, et al. Preparation and ionic conductivity of Li7P3S11-glass-ceramic electrolytes[J]. Journal of Non-Crystalline Solids, 2010, 356(44): 2670-2673.
[20] SEINO Y, OTA T, TAKADA K, et al. A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries[J]. Energy & Environmental Science, 2014, 7(2): 627-631.
[21] MIZUNO F, HAYASHI A, TADANAGA K, et al. High lithium ion conducting glass-ceramics in the system Li2S-P2S5[J]. Solid State Ionics, 2006, 177(26): 2721-2725.
[22] TACHEZ M, MALUGANI J P, MERCIER R, et al. Ionic conductivity of and phase transition in lithium thiophosphate Li3PS4[J]. Solid State Ionics, 1984, 14(3): 181-185.
[23] LIU Z, FU W, PAYZANT E A, et al. Anomalous high ionic conductivity of nanoporous β-Li3PS4[J]. Journal of the American Chemical Society, 2013, 135(3): 975-978.
[24] KANNO R, MURAYAMA M. Lithium ionic conductor thio-LISICON: The Li2S-GeS2-P2S5system[J]. Journal of the Electrochemical Society, 2001, 148(7): A742-A746.
[25] KAMAYA N, HOMMA K, YAMAKAWA Y, et al. A lithium superionic conductor[J]. Nature Materials, 2011, 10(9): 682-686.
[26] BRON P, JOHANSSON S, ZICK K, et al. Li10SnP2S12: An affordable lithium superionic conductor[J]. Journal of the American Chemical Society, 2013, 135(42): 15694-15697.
[27] KUHN A, GERBIG O, ZHU C, et al. A new ultrafast superionic Li-conductor: Ion dynamics in Li11Si2PS12and comparison with other tetragonal LGPS-type electrolytes[J]. Physical Chemistry Chemical Physics, 2014, 16(28): 14669-14674.
[28] RANGASAMY E, LIU Z, GOBET M, et al. An iodide-based Li7P2S8I superionic conductor[J]. Journal of the American Chemical Society, 2015, 137(4): 1384-1387.
[29] TATSUMISAGO M, NAGAO M, HAYASHI A. Recent development of sulfide solid electrolytes and interfacial modification for all-solid-state rechargeable lithium batteries[J]. Journal of Asian Ceramic Societies, 2013, 1(1): 17-25.
[30] KANNO R, HATA T, KAWAMOTO Y, et al. Synthesis of a new lithium ionic conductor, thio-LISICON-lithium germanium sulfide system[J]. Solid State Ionics, 2000, 130(1): 97-104.
[31] WANG Y, RICHARDS W D, ONG S P, et al. Design principles for solid-state lithium superionic conductors[J]. Nature Materials, 2015, doi: 10.1038/NMAT4369.
[32] MO Y, ONG S P, CEDER G. First principles study of the Li10GeP2S12lithium super ionic conductor material[J]. Chemistry of Materials, 2011, 24(1): 15-17.
[33] ONG S P, MO Y, RICHARDS W D, et al. Phase stability, electrochemical stability and ionic conductivity of the Li10±1MP2X12(M= Ge, Si, Sn, Al or P, and X= O, S or Se) family of superionic conductors[J]. Energy & Environmental Science, 2013, 6(1): 148-156.
[34] MERCIER R, MALUGANI J P, FAHYS B, et al. Superionic conduction in LI2S-P2S5-LiI-glasses[J]. Solid State Ionics, 1981, 5(10): 663-666.
[35] NAGATA H, CHIKUSA Y. Activation of sulfur active material in an all-solid-state lithium-sulfur battery[J]. Journal of Power Sources, 2014, 263: 141-144.
[36] AGOSTINI M, AIHARA Y, YAMADA T, et al. A lithium-sulfur battery using a solid, glass-type P2S5-Li2S electrolyte[J]. Solid State Ionics, 2013, 244: 48-51.
[37] NAGAO M, HAYASHI A, TATSUMISAGO M. High-capacity Li2S-nanocarbon composite electrode for all-solid-state rechargeable lithium batteries[J]. Journal of Materials Chemistry, 2012, 22(19): 10015-10020.
[38] NAGAO M, HAYASHI A, TATSUMISAGO M. Sulfur-carbon composite electrode for all-solid-state Li/S battery with Li2S-P2S5solid electrolyte[J]. Electrochimica Acta, 2011, 56(17): 6055-6059.
[39] NAGAO M, HAYASHI A, TATSUMISAGO M. Electrochemical performance of all-solid-state Li/S batteries with sulfu-based composite electrodes prepared by mechanical milling at high temperature[J]. Energy Technology, 2013, 1(2/3): 186-192.
[40] YAMADA T, ITO S, OMODA R, et al. All solid-state lithium-sulfur battery using a glass-type P2S5-Li2S electrolyte: Benefits on anode kinetics[J]. Journal of the Electrochemical Society, 2015, 162(4): A646-A651.
[41] LIN Z, LIU Z, FU W, et al. Lithium polysulfidophosphates: A family of lithium-conducting sulfur-rich compounds for lithium-sulfur batteries[J]. Angewandte Chemie, 2013, 125(29): 7608-7611.
[42] NAGATA H, CHIKUSA Y. An all-solid-state lithium-sulfur battery using two solid electrolytes having different functions[J]. Journal of Power Sources, 2016, 329: 268-272.
[43] CHEN M, ADAMS S. High performance all-solid-state lithium/sulfur batteries using lithium argyrodite electrolyte[J]. Journal of Solid State Electrochemistry, 2015, 19(3): 697-702.
[44] HAN F, YUE J, FAN X, et al. High-performance all-solid-state lithium-sulfur battery enabled by a mixed-conductive Li2S nanocomposite[J]. Nano Letters, 2016, 16(7): 4521-4527.
[45] KOBAYASHI T, IMADE Y, SHISHIHARA D, et al. All solid-state battery with sulfur electrode and thio-LISICON electrolyte[J]. Journal of Power Sources, 2008, 182(2): 621-625.
[46] NAGAO M, IMADE Y, NARISAWA H, et al. All-solid-state Li-sulfur batteries with mesoporous electrode and thio-LISICON solid electrolyte[J]. Journal of Power Sources, 2013, 222: 237-242.
[47] NAGAO M, SUZUKI K, IMADE Y, et al. All-solid-state lithium-sulfur batteries with three-dimensional mesoporous electrode structures[J]. Journal of Power Sources, 2016, 330: 120-126.
[48] LIN Z, LIU Z, DUDNEY N J, et al. Lithium superionic sulfide cathode for all-solid lithium-sulfur batteries[J]. ACS Nano, 2013, 7(3): 2829-2833.
[49] WHITTINGHAM M S. Electrical energy storage and intercalation chemistry[J]. Science, 1976, 192(4244): 1126-1127.
[50] AURBACH D, ZINIGRAD E, TELLER H, et al. Attempts to improve the behavior of Li electrodes in rechargeable lithium batteries[J]. Journal of the Electrochemical Society, 2002, 149(10): A1267-A1277.
[51] CHENG X B, ZHANG R, ZHAO C Z, et al. A review of solid electrolyte interphases on lithium metal anode[J]. Advanced Science, 2015, 3, doi: 10.1002/advs.201500213.
[52] HAYASHI A, HAMA S, MIZUNO F, et al. Characterization of Li2S-P2S5glass-ceramics as a solid electrolyte for lithium secondary batteries[J]. Solid State Ionics, 2004, 175(1): 683-686.
[53] NAGAO M, HAYASHI A, TATSUMISAGO M. Fabrication of favorable interface between sulfide solid electrolyte and Li metal electrode for bulk-type solid-state Li/S battery[J]. Electrochemistry Communications, 2012, 22: 177-180.
[54] CHENG X B, YAN C, PENG H J, et al. Sulfurized solid electrolyte interphases with a rapid Li+diffusion on dendrite-free Li metal anodes[J]. Energy Storage Materials,2017, doi: 10.1016/j.ensm.2017. 03.008.
Review on solid state lithium-sulfur batteries with sulfide solid electrolytes
SUN Yingzhi1, 2, HUANG Jiaqi1, 3, ZHANG Xueqiang1,ZHANG Qiang1
(1Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China;2School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China;3Advanced Research Institute for Multidisciplinary Science, School of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, China)
The development of electronic devices, such as portable electronics, electric vehicles and smart grids, brings new requirements and challenges to electrochemical energy storage system. Solid state lithium-sulfur batteries have attracted much attention for their high theoretical energy density and safety properties. Owning to high ionic conductivity and excellent interface contact, sulfide solid electrolytes are suitable for solid state lithium-sulfur batteries. This review summarizes the recent progress of solid state lithium-sulfur batteries with sulfide solid electrolytes. It focuses on the properties of sulfide solid electrolytes, the formation of efficient carrier pathways on the cathode side, and the stable lithium/electrolyte interfaces on the anode side. The future development directions of solid state lithium-sulfur batteries with sulfide solid electrolytes are also prospected.
lithium sulfur battery; solid electrolyte; ionic conductivity; sulfide; lithium metal anode
10.12028/j.issn.2095-4239.2017.0033
TM 912
A
2095-4239(2017)03-464-15
2017-03-29;
2017-04-01。
国家重点研发计划(2016YFA0202500),国家自然科学基金项目(21676160),清华大学自主科研计划。
孙滢智(1995—),男,在读本科生,主要从事高性能锂硫电池研究,E-mail:sun-yz13@mails.tsinghua.edu.cn;
张强,研究员,主要研究方向为能源材料,尤其是金属锂、锂硫电池和电催化剂,E-mail:zhang-qiang@mails.tsinghua.edu.cn。
