第一性原理计算在锂硫电池中的应用进展评述
2017-10-14侯廷政彭翃杰程新兵黄佳琦
陈 翔,侯廷政,2,彭翃杰,程新兵,黄佳琦,张 强
第一性原理计算在锂硫电池中的应用进展评述
陈 翔1,侯廷政1,2,彭翃杰1,程新兵1,黄佳琦3,张 强1
(1清华大学化学工程系绿色反应工程与工艺北京市重点实验室,北京 100084;2加州大学伯克利分校材料学院,美国加州伯克利94720;3北京理工大学前沿交叉科学研究院,北京100081)
与传统的锂离子电池相比,锂硫电池具有高能量密度而被认为是一种理想的下一代储能器件。但多硫化物的“穿梭效应”、电解质的不稳定、金属锂枝晶的生长等问题严重阻碍了锂硫电池的实际应用。第一性原理计算等理论研究方法的兴起,促进了对锂硫电池关键科学问题的理解及其实用化进程。本文基于密度泛函理论、Hartree-Fock方法、从头算分子动力学模拟等广泛应用的理论方法,综述了第一性原理计算在锂硫电池正极、电解质、负极中的具体应用以及对解决锂硫电池关键科学问题的作用,并阐述了其未来在锂硫电池研究领域的发展空间。
第一性原理计算;锂硫电池;硫正极;电解质;锂负极
从远古的青铜时代到如今硅驱动的信息时代,人类社会文明的发展与材料、化学等学科的发展息息相关。在某种程度上而言,文明的定义本身就与其是否掌握了一种新材料的制造与使用紧密相 关[1]。20世纪以来,随着计算机技术的逐渐发展,化学、材料学研究不仅仅局限于传统的实验试错法,计算机模拟已经成为了化学、材料学研究中一个重要的组成部分。通过计算机模拟,我们可以在微观层 面上理解化学物种的性质、分子间的相互作用力,在介观甚至宏观层面上理解材料的形成过程[图1(a)]。
1998年诺贝尔化学奖颁给了WALTER KOHN教授,以表彰他在量子化学——密度泛函理论(density functional theory,DFT)领域中所做出的杰出贡献。密度泛函理论的建立,使得量子化学在实际复杂体系中的应用成为了可能。另一方面,基于密度泛函理论等量子化学方法与传统的分子动力学模拟相结合发展而来的从头算分子动力学模拟方法(molecular dynamics,AIMD)为计算材料学的研究提供了机遇与可能。统计和密度泛函或第一性原理计算相关的文献也发现,20世纪90年代以来,特别是2000年之后,发表的相关文献数量增长迅猛[图1(b)]。2011年,美国奥巴马政府宣布启动“材料基因组”计划,以缩短新材料的研发周期,降低新材料的开发成本。这一计划的实施,在全球范围内推动了计算机模拟在化学、材料学研究中的应用。
量子化学、计算材料学的迅速发展也极大地促进了能源材料相关领域的研究与发展。锂硫电池,由于具有极高的能量密度(2600 kW·h/kg),是一种理想的下一代储能器件,受到了广泛的关注[2]。然而,锂硫电池目前在实用化的进程中依然面临着许多尚未解决的基础科学问题,阻碍了其进一步的发展。如正极中活性物质多硫化物向电解液中溢出引起的“穿梭效应”、硫分子及其还原产物(Li2S和Li2S2)较差的导电能力、电解液的不稳定性、负极锂枝晶的生长等[3-8]。针对以上问题,研究者们在实验层面上进行了许多努力,如采用S/C复合正极,解决硫正极导电能力差的问题[9];采用固态电解质,解决电解液稳定性不足的问题[10];构建三维导电骨架,诱导金属锂的可控形核,从而抑制锂枝晶的生长[11-12]。但是,由于锂硫电池中发生的是多电子氧化还原过程,体系十分复杂,在实验层面上采用传统的“试错”方法很难准确地处理处于平衡态与非平衡态的锂硫电池内部的物理化学信息及其演变规律,从本质上解决上述问题。但是另一方面,随着理论化学和计算机技术的发展,第一性原理计算方法逐渐发展成为化学、物理学、材料科学、化学工程等众多学科领域中一项重要的研究方法与工具,并取得了十分广泛的应用成果。这一新兴的科学方法能够帮助我们在电子及其相互作用的微观层面上理解分子之间的相互作用,化学反应的具体过程,分子在表界面的吸附行为等。因此,这一方法能够帮助我们增进对锂硫电池现有科学问题的理解,解决其在实用化进程中所遇到的科学、技术问题,从而为设计出更加高效、安全的实用化锂硫电池提供理论指导。
本文首先简要介绍第一性原理计算方法的相关理论基础,然后在此基础上详细讨论这一方法在锂硫电池的正极、电解液、负极研究中的应用,最后对这一方法未来在锂硫电池中的可能应用进行 展望。
1 第一性原理理论基础
第一性原理计算,广义上指所有基于量子力学原理,以量子力学为基础的计算。在构建物质模型时,可以把物质看做是由分子组成的,分子可以拆分成原子,原子又可以拆分成原子核和电子。如果把固体看成是位于平衡位置的原子核和周围所包含的电子组成的,那么人们就可以不使用经验参数,通过求解Schrödinger方程来完成对电子结构的计算。常用的第一性原理计算方法有:Hartree-Fork(HF)自洽场方法、密度泛函理论(DFT)、准粒子(GW)近似、Car-Parrinello方法、从头算分子动力学(AIMD)、量子蒙特卡洛方法(QMC)等[13-16]。由于HF、DFT、AIMD这3种方法在实际中应用最为广泛,下文将对这3种方法的基本理论基础进行简单介绍。
1.1 HF方法
HF方法由HARTREE和FOCK等于20世纪二三十年代提出,是在定态薛定谔方程的基础之上,逐步采用HARTREE近似、FOCK近似、LCAO-MO近似等近似,将不可解的多粒子薛定谔方程逐步化简为可求解的单粒子薛定谔方程。
1.1.1 HARTREE近似
个粒子的薛定谔方程为
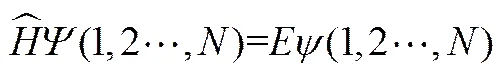
其中
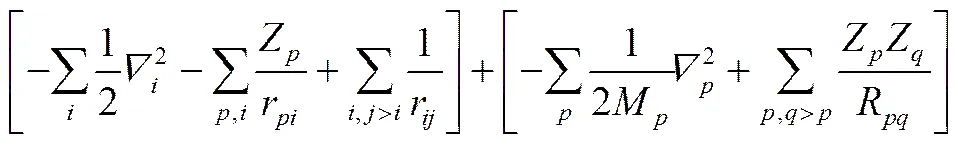
HARTREE于1928年在非相对论近似和BORN- OPPENHEIMER近似的基础上提出轨道近似(即HARTREE近似),将电子间的库仑排斥作用平均化,视每个电子在核库仑场与其它单子对该电子作用的平均场相叠加而成的势场中运动,从而单个电子的运动特性只取决于其它电子的平均密度分布(即电子云),而与其它电子的瞬时位置无关。HARTREE近似实质上是实现了电子运动与原子核运动的分离。
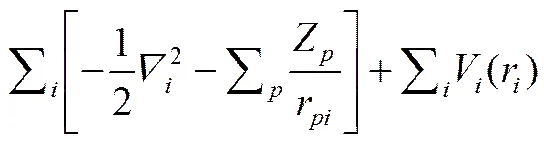
(4)
若定义单电子哈密顿算符和双电子算符:

(6)
则总的电子哈密顿算符可以简记为:
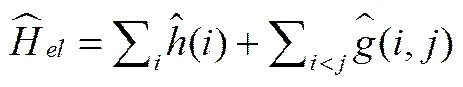
在此近似下,电子体系的波函数可以写成个电子波函数的乘积:
(8)
1.1.2 FOCK近似
HARTREE近似将多电子薛定谔方程化简为可求解的单电子薛定谔方程,但这一近似处理却使体系不满足PAULI不相容原理,即任意交换两个电子的位置,波函数反向,但不改变体系的状态。
FOCK在HARTREE近似的基础之上,采用久期行列式来描述多电子体系波函数,使其满足PAULI不相容原理要求的反对称性要求(即对公式8进行了修正)。
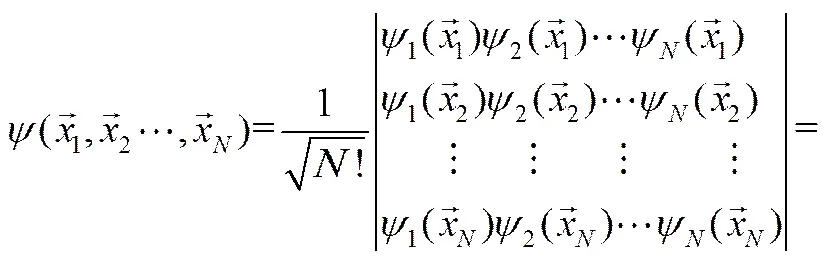
(9)
由此,并得到HARTREE-FOCK方程:
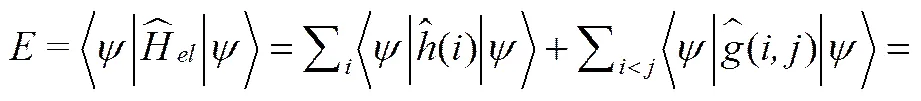
(10)
1.1.3 LCAO-MO近似
基于分子轨道的电子总能量表达式虽然形式简洁、物理意义明确,但是对于分子轨道波函数的具体形式未给出任何限制或约定,无法采用变分处理,因此必须进一步采用LCAO-MO(molecular orbitals expressed as linear combination of atomic orbitals)近似。这一近似对分子轨道进行线性展开,用一组对分子轨道集合构成数学完备集的基函数表示,即:
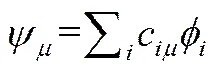
在此近似基础之上,根据能量最小原理,采用变分法确定分子轨道表示中的组合系数。从而最终可以得到著名的HARTREE-FOCK-ROOTHAAN方程:
(12)
式中,为单电子哈密顿矩阵,为密度矩阵,为双电子作用矩阵,其具体表达形式分别如下:
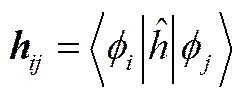
(14)
(15)
对于式(12)所示能量方程,可以采用自洽场迭代的方法进行求解,为实际求解多电子体系的薛定谔方程提供了理论可能。
1.2 密度泛函理论
DFT理论的发展历史,最早可以追溯到20世纪20年代。1927年,THOMAS和FERMI分别独立地建立了THOMAS-FERMI模型,即将体系能量表示为电子密度函数的初步模型。1964年,KOHN等[13]提出HOHENBERG-KOHN定理,严格证明了电子密度与分子能量之间的确切关系。该定理主要由HK存在定理和HK变分定理两部分组成。
1.2.1 HK存在定理
HK存在定理表述为电子密度函数唯一决定体系的外势场,从而决定体系基态的哈密顿量及一切性质。
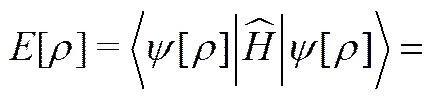
(16)
(17)
式中,是体系的总能量,是体系的电子总数,是电子的空间密度函数。由此我们可以得知,电子空间密度函数唯一决定体系基态的哈密度量及一切性质(包括体系的基态能量)。公式中表示体系的动能,表示体系的库仑能,xc表示体系的电子交换关联能,将在KOHN-SHAM方程中给出具体形式。
1.2.2 HK变分定理
HK变分定理表述为体系的能量服从能量变分原理。即:对任意的,且,则基态能量。进而可以得到,基态的能量密度应满足
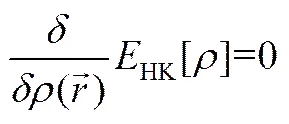
1965年,KOHN和SHAM[17]在HOHENBERG- KOHN定理的基础上建立了KOHN-SHAM方程,使密度泛函的实际应用奠定了理论基础。
(19)
式中,为体系的动能,表达式为
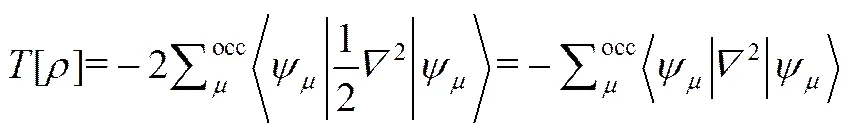
是静电相互作用能,分为原子核与电子之间的静电相互作用能V和电子与电子之间的相互作用能V。
(21)
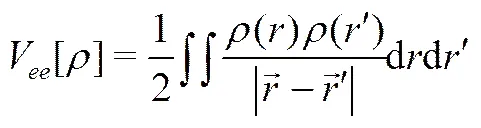
从而有
(23)
xc是电子交换关联能,是未知的,但可以用交换关联函数来表示:
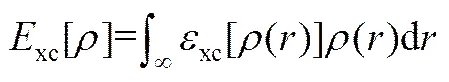
是体系的总能量,表达形式如下:
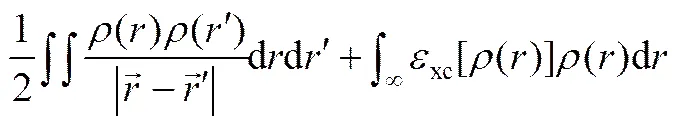
根据以上能量表达形式,结合哈密顿函数,得以得到如下的KOHN-SHAM方程:
(26)
式中,
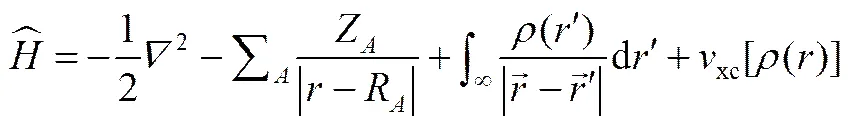
从以上介绍可以得知,将密度泛函理论应用于实际中最大的困难在于交换关联函数是未知的。如果给交换关联函数赋予某一已知的形式,那么对于某一体系(电子密度函数是确定的),体系包括能量在内的一切性质都是可以求解的。因此,发展实用而不失精度的交换关联函数是DFT理论研究中一个十分重要的领域。截止目前,DFT理论已经发展了若干交换关联函数,主要可以分为LDA(local-density approximation,局域密度近似)、LSDA(local spin-density approximation,局域自旋密度近似)、GGA(generalized gradient approximations,广义梯度近似)、Meta-GGA、Hybrid GGA、Hybrid meta-GGA。其中LDA、GGA和杂化泛函中的B3LYP在实际中被广泛地采用。
LDA泛函采用均匀电子气体系的交换关联函数作为近似,即
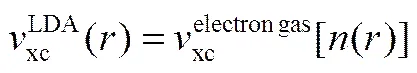
对于许多体相材料而言,其价电荷密度变化缓慢,因而较好的满足LDA的近似条件。但是对于原子簇或分子体系,这一近似假设往往不成立,需要使用更高精度的泛函。GGA泛函[18-20]考虑到了电子密度函数变化的梯度,相对于LDA在大多数情形下更为精确,其表达形式为
(29)
GGA泛函种类很多,其中最常使用的两个非经验的GGA泛函是Perdew-Wang 91(PW91)泛函[20]和Perdew-Burke-Ernzerhof(PBE)泛函[21]。杂化泛函B3LYP[22]使用精确的交换能和GGA交换泛函的混合型是来描述交换能,其具体形式如下
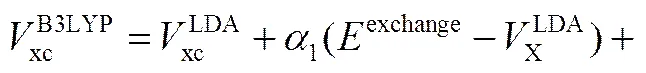
(30)
1.3 从头算分子动力学模拟
DFT和HF方法的发展也催生了AIMD[25]的发展。即使在很低的温度下,分子和原子总是不断运动的,这一运动过程可以将分子、原子视为点,采用经典的牛顿力学、经验势函数的方法进行描述,这一基本思想便是分子动力学模拟的想法。如果使用量子力学描述体系中电子和原子核的状态,将经典的牛顿动力学扩展到薛定谔方程,那么分子动力学模拟将变得更加微妙,这也便是从头算分子动力学的基本想法。
为了避免求解含时的薛定谔方程,从头算分子动力学模拟在实际使用中采用了一定的简化,其中一种最常用的方法是仅考虑基态的波函数,并且假定波函数总是取得极小值。这一方法被称作BORN- OPPENHEIMER分子动力学模拟(BOMD),其具体数学表达式如下
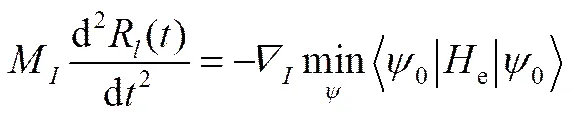
(32)
(33)
式中,R是原子的位置,Z是原子所带电荷数,M是原子质量,r是电子位置,e是电子质量。
在得到AIMD的具体数学表达式后,下一步便是求解薛定谔方程,其求解方法中大家普遍感兴趣的是HARTREE-FOCK方法和DFT。
在求解了薛定谔方程的基础上可以进一步求解分子动力学模拟,主要有两种方法:直接使用BORN-OPPENHEIMER方程[式(31)];CAR- PARRINELLO方法(CPMD)。前者的主要缺点在于每一步的波函数都需要进行优化。
求解了薛定谔方程之后,便需要考虑体系中原子在力的作用下发生的运动。作用在原子上的力可以如式(34)计算:
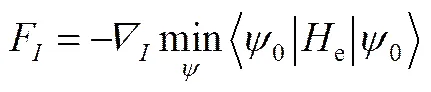
如果波函数对原子的位置没有很强的依赖关系,那么作用在原子上的力的计算可以简化为
(35)
为了避免BOMD方法的缺陷,CPMD方法主要采用了绝热近似。由于电子的质量远远小于原子核的质量,那么电子将随着原子运动,这也意味着进行复杂的波函数优化是不必要的。另一方面,单个粒子的轨道必须满足正交性约束条件,而正交性约束条件可以采用拉格朗日乘子()的方法[24],从而得到CPMD动态方程如下
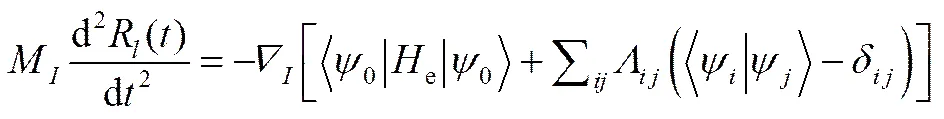
(37)
2 硫正极
锂硫电池正极采用硫作为活性物质。在放电过程中,硫分子逐渐被还原,最终生成Li2S2/Li2S。硫正极在实际中面临的主要问题[16]有:首先,硫以及不溶的Li2S2和Li2S的电子导率和离子导率均很差,导致电阻很大,从而使得在充放电过程会产生很强的极化和不可逆的能量损耗,因而,需要向其中添加能够导电的物质以改善其导电性,但这会降低其能量密度;第二,正极在循环中有很大的体积变化。由于S8(2.07 g/cm3)与Li2S(1.66 g/cm3)的密度不同,硫反应生成硫化锂的过程中会有高达79%的体积变化,这会导致正极的结构和形貌发生改变,使含硫物质与导电骨架脱离,造成容量的不可逆损失;第三,电化学反应的中间产物——多硫化锂会溶于有机电解液中。尽管溶解有利于反应更充分进行,但如若没有限制,高阶多硫化物就会扩散到负极与金属锂直接反应生成低阶多硫化物,然后进一步迁移回正极。这种所谓的多硫化物“穿梭效应”,不但会造成负极的腐蚀,破坏固体电解质界面膜,还消耗了含硫活性物质,造成容量的损失。第四,放电的最终产物是不溶且绝缘的Li2S2和Li2S,它们会覆盖在电极表面,阻碍活性物质充分有效的利用。另一方面,它们也会脱离导电骨架,使得它们无法在充电时得到还原,降低了电池的可逆性。
针对以上问题,研究者们提出了构建碳材料骨架,改善硫正极导电性能;采用金属化合物添加剂,作为宿主材料吸附多硫活性中间产物等方法。下文将主要介绍这两个方面以及充放电机理方面的理论研究工作。
2.1 碳材料骨架
自从2009年NAZAR等[26]报道采用介孔碳来改善锂硫电池的循环性能,锂硫电池的研究工作不断取得突破和进展。为了解决硫导电性差的问题,有学者采用导电骨架作为载体制备出具有纳米结构的复合正极材料,并且有效提高了硫的利用率[27-28](图2)。多孔碳[29]、碳纳米管(CNTs)[9; 29]、碳纤维[30]、石墨烯[31]、氧化石墨烯[32]、导电聚合物[33]及它们的杂化物[34-35]等多种材料都可以作为导电骨架。作为硫或硫化锂的载体,导电骨架材料有效提高了正极的比能量密度。但是,常规碳材料体系对于多硫化物的吸附能力有限。充放电过程中,多硫化物会溶解在电解液中,穿过隔膜,扩散至负极,与金属锂反应,造成不可逆的容量损失。针对这一问题,对碳材料进行杂原子掺杂,增强其多硫化物的相互作用,是一种最常规且容易实现的方法。对于碳材料的理论研究,也主要集中于碳材料及杂原子掺杂碳材料对多硫化物的吸附行为的研究。
SONG等[36]对含氮介孔碳正极材料进行了理论研究。他们认为,由于氮的掺杂,硫单质与碳材料表面发生化学键合作用,基于这个观点,他们制成了具有高面容量密度的锂硫电池正极。CHEN等[37]分析了S8、Li2S8、Li2S6、Li2S4等的结构、对称性、前线轨道能级以及其在未掺杂和氮掺杂的介孔碳表面的吸附行为,发现相对于石墨氮、吡啶氮和吡咯氮更加能显著增强介孔碳吸附多硫化物的能力。
除了多孔碳材料,碳纳米管(carbon nanotube,CNT)、石墨烯(graphene)等热门碳材料也被广泛用于锂硫电池正极骨架。对于碳纳米管体系,XI等[38]采用DFT方法计算单点能,初步探索了S8、Li2S4和Li2S等在碳纳米管表面的吸附行为。WANG等[39]采用第一性原理计算的方法研究氮掺杂碳纳米管,发现氮掺杂能有效降低锂原子沿着碳纳米管和穿过碳纳米管的能垒,同时能与硫之间形成N—S键,从而对多硫化物有强吸附的作用。基于这一结果,作者预测氮掺杂碳纳米管是一种良好的锂硫电池正极材料。TAO等[40]则报道了用于锂硫电池正极的氧、硼共掺杂的多壁碳纳米管。通过DFT计算,作者研究团队发现氧、硼共掺杂的碳纳米管对多硫化物的吸附作用要强于氧或硼掺杂以及未掺杂的碳纳米管,从而说明多种杂原子掺杂之间存在着十分显著的协同效应。此外,作者还通过可视化吸附实验证实了氧、硼共掺杂碳纳米管对多硫化物的强吸附作用。LEE等[41]利用-相互作用在碳纳米管表面诱导共价有机骨架(covalent organic framework, COF)的自组装,合成三维杂化碳材料,用于锂硫电池正极。碳纳米管骨架提供电子、离子通道;COF提供多硫化物吸附位点。CNT与COF之间的协同效应有利于LI2S的沉积/氧化,促进多硫化物的液固转化。实验结果也表明,采用COF-CNT多级骨架结构正极,能显著提升锂硫电池的性能,2.0 C下循环300圈,能保持84%的容量,远高于使用常规正极电池的15%。
相比于碳纳米管,石墨烯在锂硫电池正极中的应用更加广泛。同样,由于未掺杂的碳纳米管对多硫化物的吸附作用有限,不能有效抑制穿梭效应,需要对石墨烯材料进行掺杂改性。HOU等[42]系统地比较了各种不同氮掺杂构型(石墨氮、吡啶氮和吡咯氮、边缘掺杂和体相掺杂)的石墨烯纳米条带(锯齿型和手扶椅型)对不同多硫化物的吸附[图3(a)~(b)],发现氮倾向于在石墨烯边缘掺杂,其中吡啶、吡咯氮对多硫化物分子有较强的吸附作用,且这种吸附作用受到边缘效应、空间位阻等多种因素的作用。RAO等[43]、YIN等[44]及WANG等[45]也研究了氮掺杂石墨烯对多硫化物的吸附作用,同样发现吡啶和吡咯氮具有较强的吸附多硫化物的作用,得到了相类似的结论。此外,LI等[46]和WANG等[47]研究了氧化石墨烯/含氧官能团石墨烯对多硫化物的吸附作用[图3(c)];ZHAO等[48]研究了氮、硼共掺杂石墨烯对多硫化物的吸附作用[图3(d)];GU等[49]研究了氮、磷共掺杂石墨烯对多硫化物的吸附作用。WANG等[50]研究了一系列含有碳、氧、氮和磷原子官能团的石墨烯材料,认为这些官能团的存在可以提高对多硫化物的吸附性能。共掺杂的理论研究大多发现,共掺杂的杂原子之间存在一定的协同作用,进一步增强掺杂石墨烯对多硫化物的吸附作用(相对于单原子掺杂)。除了杂原子掺杂,RAO等[43]和LIANG等[51]还研究了石墨烯及杂原子掺杂石墨烯中的缺陷对多硫化物的吸附作用,同样发现缺陷具有增强吸附多硫化物的作用。DING 等[52]发现电解液中的硝酸根离子能增强石墨烯骨架对多硫化物的吸附作用,促进多硫化物的氧化,从而可以有效抑制穿梭效应。以上研究指出了某种掺杂或者共掺杂能够增强石墨烯骨架对多硫化物的吸附作用,并在一定层面指出杂原子掺杂能够增强对多硫化物吸附作用的化学本质。如对于氮原子掺杂,有石墨氮、吡啶氮、吡咯氮等多种掺杂形式,而其中以吡咯氮和吡咯的作用最为突出;相反,石墨氮会起到相反的效果。这是由于氮掺杂石墨烯对多硫化物的吸附作用受到杂原子及其邻近碳原子的电子形态、空间位阻等多方面因素的影响。吡咯氮和吡咯氮中拥有一对孤对电子,是路易斯碱,与多硫化物中缺电子的锂刚好可以形成较强的路易斯酸碱相互作用[53]。但是,另一方面,这些工作却没解释不同的杂原子掺杂会有不同的原因,以及如何进行用于锂硫电池正极的掺杂石墨烯的理性设计。
ZHANG等[53]系统地研究了B、N、O、P、S、Cl等元素掺杂石墨烯吸附多硫化物(Li2S、Li2S4、Li2S6、Li2S8)以及S8分子,抑制“穿梭效应”的效果,并得到了杂原子掺杂石墨烯理性设计的火山型曲线,提出了用于锂硫电池正极掺杂石墨烯理性设计原则(图4)。①杂原子应具有一对孤对电子,从而与多硫化锂中的锂之间形成路易斯酸碱相互作用;②杂原子的电负性应高于碳原子,有利于在掺杂位点形成永久偶极矩;③杂原子半径应与锂原子相匹配,有利于形成锂与杂原子之间的成键,这种成键与氢键类似,被称作锂键。④杂原子与碳骨架之间形成共轭键,增强路易斯碱性,从而增强杂原子与多硫化物之间的偶极-偶极相互作用。⑤杂原子与石墨烯本身的键合作用应足够强,否则杂原子会与多硫化物之间发生反应。基于这一设计原则讨论,仅有氮、氧两种掺杂能够有效增强石墨烯骨架对多硫化物的锚定作用,并满足其它设计要求。基于此,作者提出了氮、氧共掺杂石墨烯的结构设计,以实现更优异的电池性能。这一预测也很快得到了其它研究者的证实。XU等[54]设计一种氮、氧共掺杂的石墨烯材料,用于锂硫电池正极。他们从理论和实验两方面证明了这种氮、氧共掺杂材料能够促进放电过程中短链多硫化物的形成,从而可以有效抑制“穿梭效应”。
2.2 金属化合物添加剂
虽然碳材料具有良好的导电性能,且通过杂原子掺杂可以有效增强碳材料对多硫化物的吸附作用,从而抑制“穿梭效应”。但是,碳材料掺杂的杂原子含量有限,因而吸附多硫化物的位点有限,且过量的杂原子掺杂会改变石墨烯等碳材料的共轭结构,显著降低导电能力。因此,杂原子掺杂石墨烯用于锂硫电池正极具有一定的局限性。而金属化合物(氧化物、硫化物、碳化物、氮化物等)一般对多硫化物有较强的吸附能力,从而可以抑制“穿梭效应”;同时,部分金属化合物还能促进多硫化物之间的转化,提升电池的倍率性能;此外,一部分金属化合物还具有良好的导电性能。因此,向碳材料中引入金属化合物添加剂是解决硫正极科学难题 的一个新思路,有望进一步提升锂硫电池的整体 性能。
TAO等[55]采用导电的TiO2n-1作为锂硫电池正极材料吸附多硫化物,通过DFT计算发现,相对于TiO2,Ti4O7表面低配位的Ti原子对多硫化物具有更强的吸附作用,从而证明金属化合物表面原子的配位环境对多硫物种的吸附具有十分显著的影响。CHEN等[56]利用原子沉积法在硫正极表面沉积一层超薄Al2O3层,诱导Li2S的沉积,发现Al2O3层不仅能诱导Li2S形核,还能对其它高阶多硫化物有强吸附的作用,从而抑制多硫化物的溶出,抑制穿梭效应。
相对于金属氧化物,金属硫化物具有更好的导电性能,而且金属化合物/碳材料复合硫正极中最关键的还是如何理解硫原子与金属原子之间的相互作用。因此,金属硫化物添加剂在锂硫电池正极材料中应用更加广泛,且更有助于加深理解硫正极的关键科学问题。YUAN等[57]不仅证实了CoS2对多硫化物有强吸附作用[Li2S4@ CoS2(111)晶面石墨烯片:1.97 eV0.34 eV],还证实了CoS2具有促进高阶多硫化物向低阶多硫化物转化的功能[图5 (a)]。因此,CoS2硫正极不仅具有高能量效率(相对于不添加CoS2的硫正极电池能量效率提高10%)、高放电容量(0.5 C的放电速率下具有1368 mA·h/g的初始放电容量),还具有优异的循环稳定性(2.0 C下循环2000圈,每圈衰减小于0.034%)。SEH等[58]将TiS2、ZrS2和VS2用于锂硫电池正极,发现这类二维层状材料对硫化物均有较强的吸附作用,并实现了优异的电池性能。特别地,TiS2@Li2S复合正极在4 C的倍率和5.3 mgLi2S/cm2的高面载量下实现了503 mA·h/gLi2S的比容量。NAZAR等[59]采用导电的Co9S8与石墨烯复合,构建硫正极骨架。通过范德华力矫正的DFT计算发现,Co9S8的(002)、(202)和(008)晶面对Li2S2均有较强的吸附作用,结合能分别为3.24 eV、4.17 eV、6.93 eV,明显大于石墨对Li2S2的结合能(0.94 eV)。作者还证实,采用负载Co9S8的硫正极的电池,实现了更好的循环稳定性(0.5 C下循环1500圈,每圈衰减小于0.045%)和硫负载量[75%(质量分数),4.5 mg/cm2的面载量]。
除了金属氧化物和硫化物,金属氮化物和碳化物也被应用于锂硫电池正极材料中。PENG等[60]采用导电的TiC与石墨烯复合,不仅可以有效抑制多硫化物的溶出,还能促进多硫化物的液-液、液-固转化[图5(b)]。SALEM等[61]通过DFT计算证明了WC和TiC对Li2S8的强吸附作用,从而可以有效抑制穿梭效应。SUN等[62]采用导电VN/石墨烯复合硫正极,有效增强了对多硫化物的吸附性能[图5(c)](结合能为3.75 eV)。由于VN导电性好,硫正极表现出极低的极化和更快的反应动力学性能。相应的锂硫电池初始容量为1471 mA·h/g,且在0.2 C下循环100圈,依然能保持85%的容量。
目前关于锂硫电池正极计算的相关工作中,绝大多数工作都是通过采用强极性材料(掺杂碳材料或金属化合物),实现对多硫化物的强吸附,进而抑制穿梭效应,并强调这种吸附作用越强越好。这一想法较为直观,易于通过理论计算证明,且与对多硫化物吸附的可视化实验相符合。但另外一方面,硫正极宿主材料对多硫化物的吸附作用理论上而言应有一个上界。对多硫化物过强的吸附,一方面可能会导致多硫化物的直接分解;另一方面,特别是对通过锂键作用于多硫化物的宿主材料而言,对多硫化物过强的吸附作用对电池充电过程是极为不利的。但是对于这一观点,很难在计算层面上界定多大的吸附能是比较合适的,多强的吸附作用是过强的,且从实验层面上进行严格验证更加困难。目前,仅有少数计算工作在这方面进行了一些探讨。BALBUENA等[63]采用DFT和AIMD方法,比较了MoS2、MoO2、Fe2O3和石墨烯对多硫化物(Li2S6和Li2S8)的吸附行为。作者发现,石墨烯的孔结构对多硫化物有很好的的限域作用,且有利于多硫化物解离为多硫离子;MoO2对多硫化物有更强的吸附作用且不会导致多硫化物的解离;虽然MoS2和Fe2O3对多硫化物有更强的吸附作用,但这些材料的反应活性太强,会导致高阶多硫化物的分解,且解离的锂离子会被牢牢地吸附在MoS2和Fe2O3表面。基于这一计算结果,作者认为锂硫电池正极宿主材料的设计需在亲硫和适中的表面活性之间寻找平衡,而不是单一地考虑对多硫化物吸附能的高低。ZHANG等[64]通过第一性原理计算,从结合能、空间结构和转移电荷等角度研究了一系列具有二维结构的过渡金属氧化物、硫化物和氯化物(V2O5、MoO3、TiS2、ZrS2、VS2、NbS2、MoS2、TiCl2、ZrCl2)对多硫化物的吸附行为。作者发现,这类二维材料对多硫化物的吸附作用强弱取决于多硫化物中硫原子与二维材料之间的电荷转移,并且这种电荷转移会降低多硫化物中硫原子与锂原子之间的电子密度,从而削弱Li—S键。因此,太强的吸附作用会导致多硫化物的分解。作者由此提出,合适的电极材料应该具有适中的吸附作用——一方面要能有效吸附多硫化物,避免其向电解液中溶解;另一方面,不能使多硫化物发生分解。TAO等[65]选取MgO、Al2O3、CeO2、La2O3和CaO等不导电金属氧化物作为硫正极添加剂,从理论计算和实验两方面研究其对锂硫电池性能的影响。作者发现,高比表面积和对多硫化物强吸附作用只是选择硫正极添加剂的先要条件,氧化物表面的扩散行为也十分重要。特别是对于不导电的金属化合物,吸附的多硫化物能够扩散至导电的碳骨架上,才能保证多硫化物的完全电化学转化,诱导Li2S的可控形核。基于此,作者提出硫正极金属氧化物添加剂选择的3条标准——强吸附性能、高比表面积和优异的表面扩散性能。
从单一强调金属化合物对多硫化物的强吸附作用,到提出表面吸附与表面扩散之间的平衡,第一性原理计算增进了对锂硫电池硫正极核心问题的认识,促进了锂硫电池的进一步发展。最近,研究者越来越认识到这类金属化合物催化功能的重要性。ZHOU等[66]选取一系列过渡金属硫化物(Ni3S2、SnS2、FeS、CoS2、VS2、TiS2)作为模型,研究决定Li2S氧化过程能垒和多硫化物吸附强弱的关键因素。作者发现,硫化物的导电性、与多硫化物之间的强相互作用、快速的锂离子传输、可控的Li2S沉积、表面还原/氧化催化性能等是降低Li2S氧化过程能垒、提升电池性能的关键因素。实际上,锂硫电池中发生一个多电子氧化还原过程。一个S8分子需要得到16个电子才能被完全还原为Li2S。而且,氧化还原过程中涉及到多种多硫离子和自由基,多硫离子、自由基之间十分复杂的相互转化反应关系,这也使得锂硫电池体系更加复杂。其中,Li2S氧化过程(Li2S沉积的逆过程)涉及到多硫物种的液-固转化,是诸多多硫化物转化反应中最重要之一。因此,考虑金属化合物添加剂的催化作用时,首先考虑这一反应是比较合适的。金属化合物对锂硫电池中其它液-液、液-固转化反应的催化作用,则有待进一步的理论探索。
在理论计算不断加深对硫正极关键科学问题理解的同时,我们也需要注意到,上述大多数工作都针对一种或几种金属化合物,研究其对多硫化物的吸附作用以及其它性质,提出了一系列用于筛选的标准。其不足之处在于,未能解释不同化合物之间差异的内在原因。最近,ZHANG等[67]选取3D过渡金属硫化物作为研究对象,考虑其对多硫化物和吸附以及其表面锂扩散行为(图6)。作者发现,过渡金属硫化物与碳材料对多硫化物的作用形式十分不同。后者通过锂键与多硫化物相互作用,而前者主要通过硫键与多硫化物相互作用。作者还得到了3D过渡金属硫化物吸附多硫化物的元素周期律型火山型曲线,这一规律一方面可以直观再现通过锂键、硫键与多硫化物作用的区别;另一方面可以很好地解释不同金属硫化物之间差异的内在原因。3D过渡金属硫化物与多硫化物之间的相互作用主要通过dp相互作用产生,因此结合能高低取决于过渡金属3d轨道的电子组态。在10组硫化物中,VS因为具有三个单电子,与多硫化物的相互作用最强,且其表面锂扩散的能垒仅有0.22 eV,甚至低于锂在石墨烯表面的扩散能垒0.31 eV。这一工作,在元素周期律的层面上解释了不同过渡金属硫化物吸附多硫化物的本质差异,提出了用于筛选硫正极宿主材料的标准,有助于锂硫电池正极的理性设计与高通量筛选。
2.3 其它材料体系
对于锂硫电池正极抑制穿梭效应的计算,除了碳材料和金属化合物添加剂两大类,还有一些有意思的工作:有的是研究一些新型的材料体系;有的则是研究黏结剂的作用;有的研究形核的Li2S对多硫化物的作用等。
ZHAO等[68]采用DFT计算,研究了磷烯对多硫化物的锚定作用以及多硫化物在磷烯表面的扩散行为。作者发现磷烯对长链多硫化物(Li2S8、Li2S6、Li2S4和Li2S3)具有适中的吸附作用,且吸附的多硫化物能够保持原始的结构;长链多硫化物在磷烯的锯齿方向扩散能垒仅为0.20 eV。基于这一计算结果,作者预测磷烯是一种潜在锂硫正极材料。SUN等[69]同样计算了磷烯对多硫化物的吸附强度,得到了类似的吸附强度,发现磷烯对多硫化物中的锂和硫原子均有吸附作用,并且在实验层面上制备了磷烯用于锂硫电池隔膜。结果表明,引入的磷烯硫正极具有更高的比容量和更好的循环稳定性。LI等[70]采用DNA分子装饰硫正极,利用DNA分子中的磷酸、氨基、亚氨基、羰基、羟基等极性基团吸附多硫化物,发现—P== O和==N—位点具有最强的吸附作用。
由于传统的黏结剂[如聚(偏二氯)乙烯,PVDF]对多硫化物不具有亲和性,WANG等[71]采用偏二氯乙烯和其它单体的共聚物作为黏结剂,通过DFT计算,比较各种黏结剂对多硫化物的吸附作用,发现聚(偏二氟乙烯-三氟乙烯)[P(VDF-TRFE)]对多硫化物具有更强吸附作用,从而能够抑制穿梭效应。SEH等[72]对含有不同官能团的黏结剂与硫化锂之间的相互作用进行了计算,并且根据计算结果选择了 结合作用最强的一种黏结剂用于锂硫电池正极,从设计上实现了择优选择,从而实现了很好的电池 性能。
实际电池体系中,隔膜与正极是紧密接触的。如果隔膜的正极侧能够有效吸附多硫化物,也可抑制多硫化物向锂负极侧扩散。ZHU等[73]计算了聚丙烯(PP)、聚丙烯腈(PAN)和SiO2对多硫化物和多硫自由基的吸附作用,发现SiO2对多硫化物和多硫自由基具有更强的吸附作用。作者还通过蒙特卡洛分子动力学模型模拟了多硫化物在PP、PAN和SiO2表面的吸附。实验结果也表明PAN/SiO2隔膜能够有效抑制多硫化物向锂负极侧扩散,与计算结果相符合。
由于锂硫电池在放电过程中,Li2S会在正极表面形核,逐渐覆盖正极表面。因此,研究Li2S表面对多硫化物的吸附作用十分重要。LIU等[74]通过计算自由能,得到放电中间产物Li2S在放电终产物Li2S表面沉积时的机理。Li2S会优先在Li2S的(110)表面而非(111)表面沉积,并且趋向于直接形成Li2S而非先形成Li2S2。
2.4 充放电机理
锂硫电池中发生的是多电子氧化还原过程,涉及复杂的电化学转化和化学转化反应。目前对锂硫电池反应机理的研究也主要集中在放电机理的研究。在实验层面上,部分研究者采用紫外-可见吸收光谱、X射线吸收光谱、液相色谱、核磁共振谱以及电化学阻抗谱等表征手段[75],指认多硫化物中间产物,并通过定量各种多硫中间产物浓度的变化,提出相应的放电机理。但是,由于多硫化物中间产物种类繁多,各个中间产物的谱学信号存在一定的相互干扰,且放电过程中涉及到十分重要但难以定量检测的多硫自由基。因此,仅仅从实验层面上研究锂硫电池的放电机理,存在较大的困难。而第一性原理计算,则为锂硫电池放电机理的研究提供了新的方法与手段。通过理论研究,可以定量多硫物种的稳定性、彼此之间相互转化过程中的能量变化,从而可以逐步理解锂硫电池的放电机理。
为了从理论上研究锂硫电池放电机理,首先需要了解S8分子及其放电产物的结构信息、理化性质。SHANG等[76]基于密度泛函理论,采用不同的泛函研究了α-S及相关硫化物的结构、弹性、声子、光学及热力学性质,并发现范德华力校正对硫及硫化物的计算十分重要。VIJAYAKUMAR等[77]研究了多硫化物的结构及稳定性,发现低阶多硫化物(如Li2S4)倾向于形成簇状结构,并在二甲基亚砜(dimethyl sulphoxide,DMSO)溶剂中有二聚的倾向;高阶多硫化物(如Li2S6和Li2S8)则倾向呈现链状结构。此外,作者还发现可以发生分解生成自由基。针对锂硫电池体系中的多硫自由基,PASCAL等[78]通过DFT计算了多硫分子、多硫自由基、多硫离子的X射线吸收光谱,指认了各种多硫物种的特征峰位置。特别地,作者指认2468 eV附近的特征峰对应自由基,但由于这一特征峰的信号较弱,在实验中很难被检测到。通过这一理论研究,作者证实了S8分子在放电过程中会生成自由基,且自由基的存在显著影响放电机理。
锂硫电池放电机理研究中一个较大的争议是S8分子放电最终产物是Li2S还是Li2S2。对于过渡金属硫化物而言,MS2(M指过渡金属)更加常见,且结构更加稳定。而在锂硫电池体系中,放电最终产物与放电机理及条件息息相关,且Li2S和Li2S2均是固相,因此从实验层面上确定最终放电产物存在较大的困难。而第一性原理计算,则可以从热力学上判断放电最终产物,并从动力学上进行相关讨论。FENG等[79]认为Li2S2是放电中间产物,只在非平衡状态下存在。SEE等[80]结合DFT计算和7Li核磁共振谱研究,认为锂硫电池放电过程中Li2S是唯一的固相产物。YANG等[81]通过研究Li2S、Li2S2和S8之间转化反应的能量变化,也认为Li2S2是放电中间产物,并会自发歧化生成Li2S。PAOLELLA等[82]通过计算认为Li2S2是由更高阶的多硫化物经过歧化反应生成的,处于非平衡状态,只在特定情形下形成,不是放电最终产物。PARK等[83]同样认为Li2S2处于介稳状态,不是最终放电产物。此外,LIU等[84]研究了Li2S2的电荷转移机理,发现Li2S2是半导体,不能传输自由电子,但可以依靠其中的缺陷(锂空位和孔极化子,hole polaron)在电极和多硫化物中间传递电荷。CHEN等[85]研究了Li2S的原子、电子结构以及稳定性,发现虽然Li2S晶体是不导电的,但是锂原子的插入/脱出会显著改变Li2S的电子结构。特别地,锂原子在Li2S (111)表面的脱出会引起Li2S纳米颗粒费米能级向表面硫原子的价态偏移,使Li2S (111)具有导电性。从以上讨论可知,大多数研究者认为Li2S是锂硫电池放电的终产物,而Li2S2可能在充放电过程中扮演着重要的中间产物的作用。但同时,我们也需要认识到,大多计算结果都是在热力学上证明了Li2S更加稳定,而电池使用过程中,动力学行为甚至更加重要;同时锂硫电池放电机理可能受到多方面因素(电解液种类、正极材料种类、放电速率等)的调控,不同体系下,放电机理存在一定的差异。也因此,对于这一争论,至今尚未完全解决。
通过理论计算研究锂硫电池的充放电机理,从根本上而言还是需要研究各种多硫化锂、多硫离子之间的相互转化反应。ASSARY等[86]首先研究了各种多硫分子、多硫化锂的电子亲和力,从而得到不同多硫物种的还原电位,并且与实验较好地吻合。在此基础之上,作者进一步研究多硫化物之间相互转化反应的反应能和吉布斯自由能变,从而判断各个多硫化物之间的相互转化在实际过程中是否可能发生(热力学判据)。基于此,作者提出了锂硫电池的放电机理(图7)。该反应机理认为、、和是最主要的反应中间产物,且在完全利用时最多的中间产物是。通过这一反应机理,我们可以认识到,低阶多硫化物在充放电过程中是相对重要的,而且自由基可能发挥着非常重要的作用。WUJCIK等[87]首先计算了各种多硫化物、多硫离子的X射线近边吸收光谱的K边光谱,再对在不同电压下测得的实验结果进行拟合,得到不同电位下各种多硫化物、多硫离子的浓度,进而推断锂硫电池的放电机理。在这一工作中,作者同样检测到了各种多硫自由基的存在(LiS3、LiS4和LiS5),并且自由基主要存在于放电早期(电位高于2.25 V),在放电末期(电位区间:1.5~2.02 V)基本上不存在。
在锂硫电池放电机理的研究中,一个普遍认同的观点是,存在自由基,且自由基很可能发挥着十分重要的作用。在各种多硫自由基中,尤其以自由基的研究最为广泛。自由基的研究最早可以追溯到19世纪70年代[88-90]。青金石中的蓝色就是被认为是自由基导致的[91]。HUNSICKER等[92]率先于19世纪90年代采用DFT计算研究了各种多硫自由基(到)的空间结构、振动频率、光电子能谱等。TOSSELL等[93]采用不同的泛函和基组(B3LYP、CCSD、CCSD(T)、cc-pVTZ、CBSB7等)研究了自由基的结构、振动频率、红外及拉曼谱学。但是目前专门针对锂硫电池体系自由基的理论研究十分缺乏,从而也限制了锂硫电池充放电机理的进一步理解。自由基的理论研究是未来锂硫电池充放电机理研究的一个重要方面。
3 电解质
在锂硫电池中,电解质起着导通锂离子,连接正负极的作用。锂硫电池电解质在实际应用中的主要问题如下。①与传统锂离子电池所不同的是,锂硫电池电解质不仅仅需要考虑其黏度、对锂盐的溶解度等因素,还需要考虑多硫化物在其中的溶解度,更需要考虑其化学、电化学稳定性。由于锂硫电池采用金属锂作为负极,且金属锂十分活泼,电解质的稳定性在锂硫电池中尤为重要。②为了提高锂硫电池安全性,采用固态电解质是一种有效途径。固态电解质目前主要面临着低离子导率和界面问题。
在锂硫电池电解质的理论研究中,液态电解液的研究着重于其稳定性研究,希望计算研究电解液不稳定性的内在原因,以筛选出性能更加优异的电解液;固态电解质的研究中着重于锂离子传输机制,以筛选出更高锂离子导率的固态电解质材料。
3.1 液态电解质
1,3-二氧戊烷(1,3-dioxolane,DOL)和二乙醇二甲醚(dimethoxyethane,DME)是目前锂硫电池中最常用的电解质有机溶剂。但DOL/DME电解液在实际使用过程中依然会发生十分显著的胀气现象,即电解液发生了分解。DOL/DME电解液为什么会分解?又是如何分解的?这些都是锂硫电池电解液研究中十分基础但十分重要的问题。CHEN 等[94]首先采用HF计算,研究了DOL、DME、碳酸亚乙酯(ethylene carbonate, EC)等常见有机溶剂的分子前线轨道,发现EC分子的最低未占据轨道(lowest unoccupied molecular orbital,LUMO)低于锂负极电极电位,从而不稳定;而DOL、DME分子的LUMO轨道略高于锂负极电极电位,具有相对稳定性。但是在实体电池体系中,由于极化等因素的存在,DOL和DME溶剂在金属锂的作用下依然会发生分解。这一推测得到了AIMD计算结果的证实。基于此,作者还提出了DOL、DME分子发生分解的反应机理。作者比较了是否存在金属锂时分解反应的能垒,发现金属锂能显著降低DOL、DME分解反应能垒,从而也证实DOL、DME是在金属锂的作用下发生分解的[图8(a)~(c)]。这一研究结果揭示了DOL、DME等常规有机溶剂不稳定的本质原因,并提出抑制锂硫电池中电解液的分解的可行方案——在金属锂表面电镀一层保护膜,将溶剂与金属锂负极隔离开。这一策略也得到了实验的 证实。
为了稳定电解液与金属锂之间的界面,一种常规的方法是向电解液中添加锂盐添加剂,如双氟磺酰亚胺锂[lithium bis(fluorosulfonyl)imide,LiFSI]、双三氟甲烷磺酰亚胺锂[lithium bis(trifluorome- thanesulfonyl)imide,LiTFSI]、二氟草酸硼酸锂(lithium oxalyldifluoroborate,LiODFB)等。WU 等[95]向DOL/DME电解液中添加LiODFB来稳定DOL、DME溶剂分子。作者通过计算发现,一方面LiOFDB可以分解生成,进而在锂负极表面形成LiF保护锂负极;另一方面,LiOFDB分解生成的硼酸盐阴离子可以与DOL、DME溶剂分子相互作用。YUSHIN等[96]通过电解液中加入LiI添加剂,在金属锂表面形成LiI层。作者通过计算提出金属锂表面形成LiI层的反应机理:首先是在3 V左右的电位下,溶液中形成碘自由基,碘自由基与DME分子发生反应,夺走DME分子中一个氢原子,生成HI,生成的HI与金属锂反应生成LiI;另一方面,被夺走H的DME自由基会发生聚合反应。作者还比较了LiI与LiTFSI添加剂的区别:前者夺走DME分子上的H的反应是放热反应,而后者则是吸热 反应。
除了添加锂盐,改善电解液与金属锂之间的界面,从而稳定电解液,寻找稳定的有机溶剂及作为电解液则是另一个研究思路。PARK等[97]以还原电位为指标,对49种常见有机溶剂进行筛选,从中筛选出了22种相对稳定(还原电位低于-0.5 V)的有机溶剂作为锂金属电池电解液。作者还通过AIMD计算所筛选的22种有机溶剂与金属锂之间的表面,由于有机溶剂通过氧官能团与金属锂作用,通过烷烃基团的修饰作用,能够增大溶剂与金属锂之间的表面能,从而稳定有机溶剂。
3.2 固态电解质
由于液态电解液一般采用有机物作为溶剂,具有可燃性,且存在显著的胀气现象,液态电解液存在一定的安全隐患。为了发展安全、可能的高能量密度电池,采用固态电解质是未来发展的方向。针对固态电解质的理论研究,在锂离子电池体系相对较多。UC Berkeley的CEDER、Maryland的MO等研究者在锂离子固态电解质理论研究中做出了许多杰出的工作,而专门针对锂硫电池体系的固态电解质理论研究很少。
在锂离子电池中研究相对较多的固态电解质主要有Li10GeP2S12(LGPS)、Li7La3Zr2O12(LLZO)等体系。对这些固态电解质的理论研究主要集中于其锂离子导率和稳定性。这也是用于锂离子电池和锂硫电池固态电解质的共性问题。MO等[98]研究了LGPS的稳定性,发现LGPS在相图中处于介稳状态,低电位下在金属锂的作用下会被还原,高电位下脱出锂会发生分解。而实际中LGPS的电化学稳定窗口高达5 V,这是由于LGPS分解生成的Li2S和P2S5覆盖在LGPS表面,能有效起到保护作用。HAN等[99]研究了LGPS和LLZO这两种典型的锂离子固态电解质的热力学电化学稳定窗口(图9),测得这两种固态电解质的电化学稳定窗口分别为1.71~2.14 V、0~3 V(Li/Li+),发现传统的实验测试方法中,由于固态电解质与电极之间的接触不充分,实验测得的电化学稳定窗口往往偏高。这一计算结果也表明,固态电解质与电极接触时分解生成中间层一方面能起到保护固态电解质的作用;另一方面也导致了较高的界面电阻,影响电池的整体性能。MIARA等[100]研究了Rb、Ta等杂原子掺杂对固态电解质LLZO的离子导率和稳定性的影响,发现少量的杂原子掺杂并不会显著改变锂离子在LLZO中的迁移路径,但会显著改变LLZO中锂离子的浓度,从而改变锂离子导率。经过优化,Li6.75La3Zr1.75Ta0.25O12具有最低的扩散能垒(19 meV)和最高的室温锂离子导率(300 K,10-2S/cm)。此外,作者还发现LLZO晶胞尺寸对锂离子导率也会有显著的影响——LLZO晶胞尺寸减小会引起锂离子导率的迅速下降,但是增大LLZO的晶胞尺寸却不会有明显的影响。因此,采用半径大的金属进行掺杂,并不一定能够显著增大LLZO的锂离子导率。ONG等[101]研究了Li(10±1)MP2X12(M = Ge、Si、Sn、Al或P、X = O、S或Se)的化学、电化学稳定性和锂离子导率,发现同/异价态阳离子取代Ge4+的影响很小(Li10SiP2S12和Li10SnP2S12与LGPS具有相类似的化学、电化学稳定性和锂离子导率);相反,阴离子取代却有十分显著的影响。如Li10MP2O12不仅不稳定,而且锂离子导率低;Li10MP2Se12的锂离子导率有所提高,但电化学稳定性却有所下降。同样,作者也探讨了晶胞尺寸对锂离子导率的影响,得到了与MIARA等同样的结论。基于以上讨论,作者认为作为固态电解质阴离子是相对较优的。基于以上的论述可以发现,在固态电解质中,阴离子与金属阳离子相比,对整体的锂离子导率和稳定性具有更加显著的影响,而且是一种相对最优的阴离子选择。那么,阴离子以什么样的空间构型分布存在最有利于锂离子的扩散。WANG等[102]考虑bcc、fcc、hcp等不同空间构型的锂离子导率,并研究锂离子在其中的扩散路径、扩散能垒、空间分布密度等,发现阴离子分布具有体心立方结构的锂离子导体最有利于锂离子的扩散。基于体心立方的阴离子骨架结构,允许锂离子在相邻的四面体之间跃迁,从而扩散能垒相对较小,有利于提高锂离子导率。这一设计原则的发现使得锂离子快导体的高通量筛选成为了可能;也与实验中发现LGPS和Li7P3S11是良好的锂离子快导体的事实相吻合,并深层次揭示了这两种材料锂离子导率高的本质原因。
4 锂负极
由于金属锂具有较高的反应活性,与电解质之间存在界面反应,形成固态电解质层(solid electrolyte interphase,SEI)。锂硫电池金属锂负极实际中面临的主要问题有:①由于原位生成的SEI机械强度较差,很难有效保护金属锂;金属会刺穿SEI层,与电解质接触,进一步反应;②金属锂在形核过程中,也会由于界面的不均匀性而导致沉积的不均匀性,从而生成锂枝晶。锂枝晶的生成,容易造成金属锂的粉化,降低库仑效率;同时可能刺穿隔膜,与正极接触,引发安全隐患。
针对锂负极存在的这些问题,理论研究可以深入研究金属锂与电解质之间的界面反应,并从理论上提出保护锂负极的可行性方案。
4.1 界面反应
针对金属锂与电解质之间的界面反应,美国德州农机大学(Texas A&M University,TAMU)的BALBUENA教授等在这方面做出了许多突出的工作。CAMACHO-FORERO等[103]采用AIMD和DFT计算方法研究了DME/LiTFSI、DOL/LiTFSI电解液与金属锂之间界面反应,发现LiTFSI优先在金属锂表面分解生成LiF,且反应十分迅速,而DME和DOL溶剂在模拟过程中相对稳定,未发生分解反应(图10)。此外,作者还研究了电解液中的多硫化物与锂负极之间的界面反应,发现无论是环状还是链状的Li2S8分子均会在金属锂表面被完全还原为Li2S。CAMACHO-FORERO等[104]还研究了LiFSI和LiTFSI盐浓度对电解液与金属锂之间界面反应的影响。作者发现高盐浓度和低盐浓度电解液之间最大的区别在于:低盐浓度电解液中,锂盐主要以反式结构存在,而在高盐浓度电解液中,锂盐主要以顺式结构存在。锂盐的顺反结构会影响其在金属锂表面的界面反应行为。而对比两种锂盐,作者发现与LiTFSI所不同的是,LiFSI会在金属锂表面完全分解,从而有效生成LiF保护层。相类似地,ZHANG等[105]报道了高盐浓度的DME电解液可以稳定金属锂负极,并且作者通过分子动力学模拟发现,高盐浓度电解液中,绝大多数阴离子都是以配位的形式存在,并且所有的锂离子都是溶剂化的。针对Li2S8分子在金属锂表面反应生成Li2S这一过程,LIU等[106]做了更加深入的理论研究,发现在Li(110)表面倾向于生成无序的Li2S,而在Li(111)表面倾向于生成完美的Li2S(111)面,并且Li2S在金属锂表面形核的过程是热力学稳定的。此外,形核的Li2S层是稳定的,与金属锂表面之间存在化学相互作用,有望成为能有效抑制锂枝晶生长的SEI层。此外BALBUENA等[107-110]还研究相关电解液溶剂/添加剂[EC、DEC(diethyl carbonate)、VC(vinylene carbonate)等]与金属锂及相关表面(Li2S、LiSi等)之间的界面反应。
4.2 锂保护策略
由于金属锂与常规电解液之间存在明显的界面反应,金属锂片很难被直接用于锂硫电池,必须采取相关的保护措施,稳定金属锂-电解质界面。目前,金属锂保护策略主要可以分为两种:向电解液中添加添加剂,改善电解液与金属锂原位生成的SEI,从而保护锂负极;在金属锂表面镀上一层保护膜,从而保护金属锂。
金属锂与电解液原位生成的SEI无机层的主要成分有LiF、LiCO3、Li2O等。其中LiF被广泛认为是一种有效的SEI组分,能有效保护金属锂。ZHANG等[111]结合DFT和AIMD计算研究了EC、DEC溶剂以及FEC(fluorinated ethylene carbonate,氟代碳酸乙烯酯)添加剂与Li、LiF之间的界面反应,发现EC和DEC分子会在金属锂表面发生分解,而在LiF表面保持稳定[图11(a)~(c)]。这一结果直接验证了LiF能够有效抑制金属锂与电解液之间的反应,从而有效保护金属锂。LEUNG等[112]对SEI组分的稳定性进行了分析,提出了3条指标:热力学稳定性、电化学稳定性和界面稳定性。两种重要的SEI无机、有机组分,Li2CO3和二碳酸亚乙酯是电化学稳定但热力学不稳定的,并且Li2CO3会在金属锂表面反应进一步生成Li2O。
除了使用电解液添加剂改善SEI成分,在金属锂表面负载一层二维材料也是一种常用的方法。锂离子在这类二维材料中的迁移行为及二维材料本身的力学性能是筛选金属锂保护层所需考虑的主要问题。UTHAISAR等[113]发现锂在石墨烯表面的扩散行为有着明显的边缘效应——锂倾向于吸附在石墨烯的边缘且在石墨烯边缘的扩散能垒比在石墨烯体相中的扩散能垒要低0.15 eV。ZHANG等[114]则系统地研究了用于金属锂负极保护的BN、石墨烯、磷烯、SnSe、SnS、硅烯、锗烯、锡烯等二维材料,发现缺陷、晶体结构、键长、键角、金属邻近效应等是显著影响保护层材料锂离子导率和力学性能的关键因素。引入缺陷、增大缺陷结构的孔半径和金属邻近作用能够显著降低锂离子穿扩二维材料的能垒,从而提高锂离子导率;但同时也会降低二维材料的刚性和硬度,从而不利于抑制锂枝晶的生长。因此,作者认为,在筛选用于金属锂保护的二维材料时,应在提高锂离子导率与抑制锂枝晶之间寻找平衡。此外,这一研究还从原子间相互作用、电荷转移的微观层面上揭示了引入缺陷能够有效降低锂离子扩散能垒的深层原因。空位缺陷附近的电子密度相对较低,从而可以显著降低锂离子穿过二维材料时,二维材料向锂离子转移的电荷,从而能够显著降低二维材料与锂离子之间的重合电荷,削弱二者之间的库仑作用力,进而有效降低锂离子的扩散能垒。而钠离子在穿过这些二维材料时具有更高的扩散能垒,一方面是由于钠离子半径更大,空间位阻大;另一方面,钠离子与二维材料相互作用时,会引起更多的电荷转移,从而钠离子与二维材料之间的相互作用力更大,扩散能垒更大。这一研究工作深层地揭示了影响锂离子穿过二维材料扩散能垒的关键因素,同时也认识到提高锂离子导率与抑制锂枝晶生长之间的必然矛盾,为金属锂负极保护层材料的高通量筛选提供了很好的借鉴。但同时我们也需认识到,作为金属锂保护层材料,其电子特性特性也是一个十分关键的因素。如果保护层材料具有较好的导电性,那么锂离子将在其表面直接沉积,从而起不到保护金属锂的效应。另一方面,引入空位缺陷能够提供锂离子扩散通道,从而降低锂离子扩散能垒,这一策略的缺点已在该研究工作中详细讨论;利用小分子组装,进行分子孔道的设计,如COF、分子筛等新型材料,则可以避免这一缺点,有望在金属锂保护中取得实际的应用。
5 总结与展望
随着计算机科学技术与密度泛函理论等的逐渐发展,第一原理计算在实际中取得了越来越来越广泛的应用,极大地促进了化学、材料、化工等相关科学的发展。一方面,计算机模拟使得一些实验上不能开展、难以开展或者危险性较大的研究成为了可能;另一方面,计算机模拟能够极大地降低研 究成本,并与实验做好结合,促进科学研究的更快发展。
在锂硫电池这一新兴领域,第一性原理计算取得一定的应用,相关总结如表1所示。在锂硫正极,一方面,从最初考虑宿主材料对多硫化物的吸附,抑制穿梭效应,到后来研究极性添加剂对多硫化物的催化作用,增进了对硫正极关键科学问题的理解以及硫正极材料的理性设计;另一方面,从研究各种多硫化物的理化性质出发,考虑多硫化物的电化学反应及之间的相互转化反应,促进了对锂硫电池充放电机理的理解。在电解质,一方面,理论计算加深了对液态电解液不稳定性及添加剂不稳定性的理解,从而为液态电解液溶剂及添加剂的筛选提供了理论指导;另一方面,理论研究深层次地揭示了锂离子在固态电解质中的传输机制,以及对固态电解质的稳定性有了更好的理解。在锂负极,理论研究加深了对锂负极-电解质界面问题的理解,基于采用保护金属的策略,为锂负极结构的理性设计提供了理论指导。
由于锂硫电池距离实用化还存在一定的距离,一些基础的科学问题还有待研究。特别是随着计算机能力的进一步增强,大规模、高精度的计算在未来越发成为可能,第一性原理计算在锂硫电池研究中有望取得更大的应用:①深入理解多硫化物与正极材料之间相互作用——锂键和硫键的化学本质;②正极材料设计的普适性规则以及高通量筛选研究;③相对于锂离子电池正极材料数据库的构建,锂硫电池正极材料数据库的构建十分缺乏,有待相关研究者的共同努力;④深入理解电池工作过程中各含硫物种的存在形态、理化性质以及与实验之间做好结合,深入理解锂硫电池充电、放电机理;⑤液态电解质的高通量筛选与理性设计;⑥固态电解质锂离子传输机理,化学、电化学稳定性研究以及高通量筛选与理性设计;⑦金属锂负极与电解质的界面反应研究;⑧金属锂保护策略的理性设计以及保护层材料的高通量筛选。

表1 第一性原理计算在锂硫电池研究中应用总结
第一性原理计算在锂硫电池中的应用,有望加深对其中关键科学问题的理解,促进相关科学技术难题的攻关,促进锂硫电池实用化进程,使高容量、高安全性、长寿命的锂硫电池早日走进人民的生活,从而提高人们的生活水平与品质,促进社会整体向前发展。
[1] U. S. Materials component strategic simulation Initiative. computational materials science: A scientific revolution about to materialize[R]. 1999.
[2] BRUCE P G, FREUNBERGER S A, HARDWICK L J, et al. Li-O2and Li-S batteries with high energy storage[J]. Nature Materials, 2012, 11(1): 19-29.
[3] HUANG J Q, ZHANG Q, WEI F. Multi-functional separator/interlayer system for high-stable lithium-sulfur batteries: Progress and prospects[J]. Energy Storage Materials, 2015, 1: 127-145.
[4] PENG H J, ZHANG Z W, HUANG J Q, et al. A cooperative interface for highly efficient lithium-sulfur batteries[J]. Advanced Materials, 2016, 28(43): 9551-9558.
[5] LIU X, HUANG J Q, ZHANG Q, et al. Nanostructured metal oxides and sulfides for lithium-sulfur batteries[J]. Advanced Materials, 2017: doi: 10.1002/adma.201601759.
[6] PENG H, CHENG X, HUANG J, et al. Review on high-loading and high-energy lithium-sulfur batteries[J]. Advanced Energy Materials, 2017, 7: 1700260.
[7] YIN Y X, XIN S, GUO Y G, et al. Lithium-sulfur batteries: Electrochemistry, materials, and prospects[J]. Angew Chem. Int. Ed. Engl., 2013, 52(50): 13186-13200.
[8] MANTHIRAM A, CHUNG S H,ZU C. Lithium-sulfur batteries: Progress and prospects[J]. Advanced Materials, 2015, 27(12): 1980-2006.
[9] PENG H J, HOU T Z, ZHANG Q, et al. Strongly coupled interfaces between a heterogeneous carbon host and a sulfur-containing guest for highly stable lithium-sulfur batteries: Mechanistic insight into capacity degradation[J]. Advanced Materials Interfaces, 2014, 1(7): 1400227.
[10] XU R, XIA X H, LI S, et al. All-solid-state lithium-sulfur batteries based on newly designed Li7P2.9Mn0.1S10.7I0.3superionic conductor[J]. Journal of Materials Chemistry A, 2017: doi: 10.1039/C1037TA01147D.
[11] ZHANG R, CHENG X B, ZHAO C Z, et al. Conductive nanostructured scaffolds render low local current density to inhibit lithium dendrite growth[J]. Advanced Materials, 2016, 28(11): 2155-2162.
[12] ZHANG R, LI N W, CHENG X B, et al. Advanced micro/nanostructures for lithium metal anodes[J]. Advanced Science, 2017, 4(3): 1600445.
[13] HOHENBERG P, KOHN W. Inhomogeneous electron gas[J]. Physical Review, 1964, 136(3B): B864-B871.
[14] FOULKES W M C, MITAS L, NEEDS R J, et al. Quantum Monte Carlo simulations of solids[J]. Reviews of Modern Physics, 2001, 73(1): 33-83.
[15] GEERLINGS P, DE PROFT F,LANGENAEKER W. Conceptual density functional theory[J]. Chemical Reviews, 2003, 103(5): 1793-1874.
[16] HOU T. The formation mechanism and design principles of the strong interface interactions in lithium-sulfur batteries[D]. Beijing: Tsinghua University, 2016.
[17] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review, 1965, 140(4A): A1133-A1138.
[18] LANGRETH D C,MEHL M J. Beyond the local-density approximation in calculations of ground-state electronic properties[J]. Physical Review B, 1983, 28(4): 1809-1834.
[19] BECKE A D. Density-functional exchange-energy approximation with correct asymptotic behavior[J]. Physical Review A, 1988, 38(6): 3098-3100.
[20] PERDEW J P, CHEVARY J A, VOSKO S H, et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation[J]. Physical Review B, 1992, 46(11): 6671-6687.
[21] PERDEW J P, BURKE K,ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[22] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange[J]. The Journal of Chemical Physics, 1993, 98(7): 5648-5652.
[23] LEE C, YANG W, PARR R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density[J]. Physical Review B, 1988, 37(2): 785-789.
[24] CHEN X. The decomposition mechanism of electrolytes in lthium-sulfur batteries: A multiscale calculation research[D]. Beijing: Tsinghua University, 2016.
[25] LAASONEN K.molecular dynamics[M]. In Biomolecular simulations: Methods and protocols. MONTICELLI, and SALONEN, eds. (Totowa, NJ: Humana Press), 2013: 29-42.
[26] JI X, LEE K T,NAZAR L F. A highly ordered nanostructured carbon-sulphur cathode for lithium-sulphur batteries[J]. Nature Materials, 2009, 8(6): 500-506.
[27] EVERS S,NAZAR L F. New approaches for high energy density lithium-sulfur battery cathodes[J]. Accounts of Chemical Research, 2013, 46(5): 1135-1143.
[28] MANTHIRAM A, FU Y, SU Y S. Challenges and prospects of lithium-sulfur batteries[J]. Accounts of Chemical Research, 2013, 46(5): 1125-1134.
[29] ZHANG S M, ZHANG Q, HUANG J Q, et al. Composite cathodes containing SWCNT@S coaxial nanocables: Facile synthesis, surface modification, and enhanced performance for Li-ion storage[J]. Particle & Particle Systems Characterization, 2013, 30(2): 158-165.
[30] ELAZARI R, SALITRA G, GARSUCH A, et al. Sulfur-impregnated activated carbon fiber cloth as a binder-free cathode for rechargeable Li-S batteries[J]. Advanced Materials, 2011, 23(47): 5641-5644.
[31] HUANG J Q, LIU X F, ZHANG Q, et al. Entrapment of sulfur in hierarchical porous graphene for lithium-sulfur batteries with high rate performance from −40 to 60℃[J]. Nano Energy, 2013, 2(2): 314-321.
[32] JI L, RAO M, ZHENG H, et al. Graphene oxide as a sulfur immobilizer in high performance lithium/sulfur cells[J]. Journal of the American Chemical Society, 2011, 133(46): 18522-18525.
[33] LI W, ZHANG Q, ZHENG G, et al. Understanding the role of different conductive polymers in improving the nanostructured sulfur cathode performance[J]. Nano Letters, 2013, 13(11): 5534-5540.
[34] PENG H J, HUANG J Q, ZHAO M Q, et al. Nanoarchitectured graphene/CNT@porous carbon with extraordinary electrical conductivity and interconnected micro/mesopores for lithium-sulfur batteries[J]. Advanced Functional Materials, 2014, 24(19): 2772-2781.
[35] ZHAO M Q, LIU X F, ZHANG Q, et al. Graphene/single-walled carbon nanotube hybrids: One-step catalytic growth and applications for high-rate Li-S batteries[J]. Acs Nano, 2012, 6(12): 10759-10769.
[36] SONG J, XU T, GORDIN M L, et al. Nitrogen-doped mesoporous carbon promoted chemical adsorption of sulfur and fabrication of high-areal-capacity sulfur cathode with exceptional cycling stability for lithium-sulfur batteries[J]. Advanced Functional Materials, 2014, 24(9): 1243-1250.
[37] CHEN J J, YUAN R M, FENG J M, et al. Conductive lewis base matrix to recover the missing link of Li2S8during the sulfur sedox cycle in Li-S battery[J]. Chemistry of Materials, 2015, 27(6): 2048-2055.
[38] XI K, CHEN B, LI H, et al. Soluble polysulphide sorption using carbon nanotube forest for enhancing cycle performance in a lithium-sulphur battery[J]. Nano Energy, 2015, 12: 538-546.
[39] WANG Z, NIU X, XIAO J, et al. First principles prediction of nitrogen-doped carbon nanotubes as a high-performance cathode for Li-S batteries[J]. Rsc Advances, 2013, 3(37): 16775-16780.
[40] JIN C, ZHANG W, ZHUANG Z, et al. Enhanced sulfide chemisorption using boron and oxygen dually doped multi-walled carbon nanotubes for advanced lithium-sulfur batteries[J]. Journal of Materials Chemistry A, 2017, 5(2): 632-640.
[41] YOO J, CHO S J, JUNG G Y, et al. COF-net on CNT-net as a molecularly designed, hierarchical porous chemical trap for polysulfides in lithium-sulfur batteries[J]. Nano Letters, 2016, 16(5): 3292-3300.
[42] HOU T Z, PENG H J, HUANG J Q, et al. The formation of strong-couple interactions between nitrogen-doped graphene and sulfur/lithium (poly)sulfides in lithium-sulfur batteries[J]. 2D Materials, 2015, 2(1): 14011.
[43] RAO D, WANG Y, ZHANG L, et al. Mechanism of polysulfide immobilization on defective graphene sheets with N-substitution[J]. Carbon, 2016, 110: 207-214.
[44] YIN L C, LIANG J, ZHOU G M, et al. Understanding the interactions between lithium polysulfides and N-doped graphene using density functional theory calculations[J]. Nano Energy, 2016, 25: 203-210.
[45] WANG Z, DONG Y, LI H, et al. Enhancing lithium-sulphur battery performance by strongly binding the discharge products on amino-functionalized reduced graphene oxide[J]. Nature Communications, 2014, 5: 5002.
[46] LI S, MOU T, REN G, et al. Confining sulfur species in cathodes of lithium-sulfur batteries: insight into nonpolar and polar matrix surfaces[J]. Acs Energy Letters, 2016, 1(2): 481-489.
[47] WANG B, ALHASSAN S M,PANTELIDES S T. Formation of large polysulfide complexes during the lithium-sulfur battery discharge[J]. Physical Review Applied, 2014, 2(3): 34004.
[48] LI F, SU Y,ZHAO J. Shuttle inhibition by chemical adsorption of lithium polysulfides in B and N co-doped graphene for Li-S batteries[J]. Physical Chemistry Chemical Physics, 2016, 18(36): 25241-25248.
[49] GU X X, TONG C J, LAI C, et al. A porous nitrogen and phosphorous dual doped graphene blocking layer for high performance Li-S batteries[J]. Journal of Materials Chemistry A, 2015, 3(32): 16670-16678.
[50] WANG X, GAO Y, WANG J, et al. Chemical adsorption: Another way to anchor polysulfides[J]. Nano Energy, 2015, 12: 810-815.
[51] LIANG Z, FAN X, SINGH D J, et al. Adsorption and diffusion of Li with S on pristine and defected graphene[J]. Physical Chemistry Chemical Physics, 2016, 18(45): 31268-31276.
[52] DING N, ZHOU L, ZHOU C, et al. Building better lithium-sulfur batteries: From LiNO3to solid oxide catalyst[J]. Scientific Reports, 2016, 6: 33154.
[53] HOU T Z, CHEN X, PENG H J, et al. Design principles for heteroatom-doped nanocarbon to achieve strong anchoring of polysulfides for lithium-sulfur batteries[J]. Small, 2016, 12(24): 3283-3291.
[54] XU N, QIAN T, LIU X, et al. Greatly suppressed shuttle effect for improved lithium sulfur battery performance through short chain intermediates[J]. Nano Letters, 2017, 17(1): 538-543.
[55] TAO X, WANG J, YING Z, et al. Strong sulfur binding with conducting Magneli-phase TiO2n-1nanomaterials for improving lithium-sulfur batteries[J]. Nano Letters, 2014, 14(9): 5288-5294.
[56] CHEN Y, LU S, ZHOU J, et al. 3D graphene framework supported Li2S coated with ultra-thin Al2O3films: Binder-free cathodes for high-performance lithium sulfur batteries[J]. Journal of Materials Chemistry A, 2017, 5(1): 102-112.
[57] YUAN Z, PENG H J, HOU T Z, et al. Powering lithium-sulfur battery performance by propelling polysulfide redox at sulfiphilic hosts[J]. Nano Letters, 2016, 16(1): 519-527.
[58] SEH Z W, YU J H, LI W, et al. Two-dimensional layered transition metal disulphides for effective encapsulation of high-capacity lithium sulphide cathodes[J]. Nature Communications, 2014, 5: 5017.
[59] PANG Q, KUNDU D,NAZAR L F. A graphene-like metallic cathode host for long-life and high-loading lithium-sulfur batteries[J]. Materials Horizons, 2016, 3(2): 130-136.
[60] PENG H J, ZHANG G, CHEN X, et al. Enhanced electrochemical kinetics on conductive polar mediators for lithium-sulfur batteries[J]. Angewandte Chemie International Edition, 2016, 55(42): 12990-12995.
[61] AL SALEM H, CHITTURI V R, BABU G, et al. Stabilizing polysulfide-shuttle in a Li-S battery using transition metal carbide nanostructures[J]. Rsc Advances, 2016, 6(111): 110301-110306.
[62] SUN Z, ZHANG J, YIN L, et al. Conductive porous vanadium nitride/graphene composite as chemical anchor of polysulfides for lithium-sulfur batteries[J]. Nature Communications, 2017, 8: 14627.
[63] KAMPHAUS E P,BALBUENA P B. Long-chain polysulfide retention at the cathode of Li-S batteries[J]. The Journal of Physical Chemistry C, 2016, 120(8): 4296-4305.
[64] ZHANG Q, WANG Y, SEH Z W, et al. Understanding the anchoring effect of two-dimensional layered materials for lithium-sulfur batteries[J]. Nano Letters, 2015, 15(6): 3780-3786.
[65] TAO X, WANG J, LIU C, et al. Balancing surface adsorption and diffusion of lithium-polysulfides on nonconductive oxides for lithium-sulfur battery design[J]. Nature Communications, 2016, 7: 11203.
[66] ZHOU G, TIAN H, JIN Y, et al. Catalytic oxidation of Li2S on the surface of metal sulfides for Li-S batteries[J]. Proceedings of the National Academy of Sciences of the United State of America, 2017, 114(5): 840-845.
[67] CHEN X, PENG H J, ZHANG R, et al. An analogous periodic law for strong anchoring of polysulfides on polar hosts in lithium sulfur batteries: S-or Li-binding on first-row transition-metal sulfides?[J]. Acs Energy Letters, 2017: 795-801.
[68] ZHAO J, YANG Y, KATIYAR R S, et al. Phosphorene as a promising anchoring material for lithium-sulfur batteries:A computational study[J]. Journal of Materials Chemistry A, 2016, 4(16): 6124-6130.
[69] SUN J, SUN Y, PASTA M, et al. Entrapment of polysulfides by a black-phosphorus-modified separator for lithium-sulfur batteries[J]. Advanced Materials, 2016, 28(44): 9797-9803.
[70] LI Q, ZHOU C, JI Z, et al. High-performance lithium/sulfur batteries by decorating CMK-3/S cathodes with DNA[J]. Journal of Materials Chemistry A, 2015, 3(14): 7241-7247.
[71] WANG H, SENCADAS V, GAO G, et al. Strong affinity of polysulfide intermediates to multi-functional binder for practical application in lithium-sulfur batteries[J]. Nano Energy, 2016, 26: 722-728.
[72] SEH Z W, ZHANG Q, LI W, et al. Stable cycling of lithium sulfide cathodes through strong affinity with a bifunctional binder[J]. Chemical Science, 2013, 4(9): 3673-3677.
[73] ZHU J, YILDIRIM E, ALY K, et al. Hierarchical multi-component nanofiber separators for lithium polysulfide capture in lithium-sulfur batteries: an experimental and molecular modeling study[J]. Journal of Materials Chemistry A, 2016, 4(35): 13572-13581.
[74] LIU Z, HUBBLE D, BALBUENA P B, et al. Adsorption of insoluble polysulfides Li2S(=1, 2) on Li2S surfaces[J]. Physical Chemistry Chemical Physics, 2015, 17(14): 9032-9039.
[75] WILD M, O'NEILL L, ZHANG T, et al. Lithium sulfur batteries, a mechanistic review[J]. Energy & Environmental Science, 2015, 8(12): 3477-3494.
[76] SHANG S, WANG Y, GUAN P, et al. Insight into structural, elastic, phonon, and thermodynamic properties of α-sulfur and energy-related sulfides: A comprehensive first-principles study[J]. Journal of Materials Chemistry A, 2015, 3(15): 8002-8014.
[77] VIJAYAKUMAR M, GOVIND N, WALTER E, et al. Molecular structure and stability of dissolved lithium polysulfide species[J]. Physical Chemistry Chemical Physics, 2014, 16(22): 10923-10932.
[78] PASCAL T A, PEMMARAJU C D,PRENDERGAST D. X-ray spectroscopy as a probe for lithium polysulfide radicals[J]. Physical Chemistry Chemical Physics, 2015, 17(12): 7743-7753.
[79] FENG Z, KIM C, VIJH A, et al. Unravelling the role of Li2S2in lithium-sulfur batteries: A first principles study of its energetic and electronic properties[J]. Journal of Power Sources, 2014, 272: 518-521.
[80] SEE K A, LESKES M, GRIFFIN J M, et al.structure search and in situ7Li NMR studies of discharge products in the Li-S battery system[J]. Journal of the American Chemical Society, 2014, 136(46): 16368-16377.
[81] YANG G, SHI S, YANG J, et al. Insight into the role of Li2S2in Li-S batteries: A first-principles study[J]. Journal of Materials Chemistry A, 2015, 3(16): 8865-8869.
[82] PAOLELLA A, ZHU W, MARCEAU H, et al. Transient existence of crystalline lithium disulfide Li2S2in a lithium-sulfur battery[J]. Journal of Power Sources, 2016, 325: 641-645.
[83] PARK H, KOH H S,SIEGEL D J. First-principles study of redox end members in lithium-sulfur batteries[J]. The Journal of Physical Chemistry C, 2015, 119(9): 4675-4683.
[84] LIU Z, BALBUENA P B,MUKHERJEE P P. Revealing charge transport mechanisms in Li2S2for Li-sulfur batteries[J]. The Journal of Physical Chemistry, 2017: 1324-1330.
[85] CHEN Y,KAGHAZCHI P. Metalization of Li2S nanoparticles in Li-S batteries[J]. Nanoscale, 2014, 6(22): 13391-13395.
[86] ASSARY R S, CURTISS L A,MOORE J S. Toward a molecular understanding of energetics in Li-S batteries using nonaqueous electrolytes: A high-level quantum chemical study[J]. The Journal of Physical Chemistry C, 2014, 118(22): 11545-11558.
[87] WUJCIK K H, PASCAL T A, PEMMARAJU C D, et al. Characterization of polysulfide radicals present in an ether-based electrolyte of a lithium-sulfur battery during initial discharge using in situ X-ray absorption spectroscopy experiments and first-principles calculations[J]. Advanced Energy Materials, 2015, 5(16): 1500285.
[88] CHIVERS T,DRUMMOND I. Characterization of the trisulfur radical anionin blue solutions of alkali polysulfides in hexamethylphosphoramide[J]. Inorganic Chemistry, 1972, 11(10): 2525-2527.
[89] LUNSFORD J H,JOHNSON D P. Electron paramagnetic resonance study offormed on magnesium oxide[J]. The Journal of Chemical Physics, 1973, 58(5): 2079-2083.
[90] CHIVERS T. Ubiquitous trisulphur radical ion.·[J]. Nature, 1974, 252: 32-33.
[91] CHIVERS T,ELDER P J. Ubiquitous trisulfur radical anion: fundamentals and applications in materials science, electrochemistry, analytical chemistry and geochemistry[J]. Chemical Society Review, 2013, 42(14): 5996-6005.
[92] HUNSICKER S, JONES R O,GANTEFÖR G. Rings and chains in sulfur cluster anions S−to: Theory (simulated annealing) and experiment (photoelectron detachment)[J]. The Journal of Chemical Physics, 1995, 102(15): 5917-5936.
[93] TOSSELL J A. Calculation of the properties of the. radical anion and its complexes with Cu+in aqueous solution[J]. Geochimica et Cosmochimica Acta, 2012, 95: 79-92.
[94] CHEN X, HOU T Z, LI B, et al. Towards stable lithium-sulfur batteries: Mechanistic insights into electrolyte decomposition on lithium metal anode[J]. Energy Storage Materials, 2017: http://dx.doi.org/10.1016/j.ensm.2017.1001.1003.
[95] WU F, QIAN J, CHEN R, et al. An effective approach to protect lithium anode and improve cycle performance for Li-S batteries[J]. Acs Applied Materials & Interfaces, 2014, 6(17): 15542-15549.
[96] WU F, LEE J T, NITTA N, et al. Lithium iodide as a promising electrolyte additive for lithium-sulfur batteries: mechanisms of performance enhancement[J]. Advanced Materials, 2015, 27(1): 101-108.
[97] PARK M S, MA S B, LEE D J, et al. A highly reversible lithium metal anode[J]. Scientific Reports, 2014, 4: 3815.
[98] MO Y, ONG S P,CEDER G. First principles study of the Li10GeP2S12lithium super ionic conductor material[J]. Chemistry of Materials, 2012, 24(1): 15-17.
[99] HAN F, ZHU Y, HE X, et al. Electrochemical stability of Li10GeP2S12and Li7La3Zr2O12solid electrolytes[J]. Advanced Energy Materials, 2016, 6(8): 1501590.
[100] MIARA L J, ONG S P, MO Y, et al. Effect of Rb and Ta doping on the ionic conductivity and stability of the garnet Li7+2x–y(La3–xRb)(Zr2–yTa)O12(0≤≤0.375, 0≤≤1) superionic conductor: A first
principles investigation[J]. Chemistry of Materials, 2013, 25(15): 3048-3055.
[101] ONG S P, MO Y, RICHARDS W D, et al. Phase stability, electrochemical stability and ionic conductivity of the Li10±1MP2X12(M = Ge, Si, Sn, Al or P, and X = O, S or Se) family of superionic conductors[J]. Energy & Environmental Science, 2013, 6(1): 148-156.
[102] WANG Y, RICHARDS W D, ONG S P, et al. Design principles for solid-state lithium superionic conductors[J]. Nature Materials, 2015, 14(10): 1026-1031.
[103] CAMACHO-FORERO L E, SMITH T W, BERTOLINI S, et al. Reactivity at the lithium-metal anode surface of lithium-sulfur batteries[J]. The Journal of Physical Chemistry C, 2015, 119(48): 26828-26839.
[104] CAMACHO-FORERO L E, SMITH T W,BALBUENA P B. Effects of high and low salt concentration in electrolytes at lithium-metal anode surfaces[J]. The Journal of Physical Chemistry C, 2017, 121(1): 182-194.
[105] QIAN J, HENDERSON W A, XU W, et al. High rate and stable cycling of lithium metal anode[J]. Nature Communications, 2015, 6: 6362.
[106] LIU Z, BERTOLINI S, BALBUENA P B, et al. Li2S film formation on lithium anode surface of Li-S batteries[J]. Acs Applied Materials & Interfaces, 2016, 8(7): 4700-4708.
[107] MA Y, MARTINEZ DE LA HOZ J M, ANGARITA I, et al. Structure and reactivity of alucone-coated films on Si and LiSisurfaces[J]. Acs Applied Materials & Interfaces, 2015, 7(22): 11948-11955.
[108] MARTINEZ DE LA HOZ J M, LEUNG K,BALBUENA P B. Reduction mechanisms of ethylene carbonate on Si anodes of lithium-ion batteries: Effects of degree of lithiation and nature of exposed surface[J]. Acs Applied Materials & Interfaces, 2013, 5(24): 13457-13465.
[109] SOTO F A, MA Y, MARTINEZ DE LA HOZ J M, et al. Formation and growth mechanisms of solid-electrolyte interphase layers in rechargeable batteries[J]. Chemistry of Materials, 2015, 27(23): 7990-8000.
[110] MARTINEZ DE LA HOZ J M,BALBUENA P B. Reduction mechanisms of additives on Si anodes of Li-ion batteries[J]. Physical Chemistry Chemical Physics, 2014, 16(32): 17091-17098.
[111] ZHANG X Q, CHENG X B, CHEN X, et al. Fluoroethylene carbonate additives to render uniform Li deposits in lithium metal batteries[J]. Advanced Functional Materials, 2017, 27(10): 1605989.
[112] LEUNG K, SOTO F, HANKINS K, et al. Stability of solid electrolyte interphase components on lithium metal and reactive anode material surfaces[J]. The Journal of Physical Chemistry C, 2016, 120(12): 6302-6313.
[113] UTHAISAR C,BARONE V. Edge effects on the characteristics of Li diffusion in graphene[J]. Nano Letters, 2010, 10(8): 2838-2842.
[114] TIAN H, SEH Z W, YAN K, et al. Theoretical investigation of 2D layered materials as protective films for lithium and sodium metal anodes[J]. Advanced Energy Materials, 2017: 1602528.
Review on the applications of first-principles calculation in lithium-sulfur batteries
1,1,2,1,1,3,1
(1Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Department of Chemical Engineering, Tsinghua University, Beijing 100084, China;2Department of Materials Science and Engineering, University of California Berkeley, Berkeley 94720, California, United States;3Advanced Research Institute for Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China)
Lithium-sulfur (Li-S) batteries are strongly considered as next-generation energy storage devices for its extremely high energy capacity compared with traditional lithium ion batteries (LIBs). However, several issues such as the shuttle of polysulfides, the instability of electrolyte, and the growth of lithium dendrites restrict their practical applications. The development of first-principles method has promoted the understanding of the key questions in Li-S batteries and their practical process. This paper reviews the applications of density functional theory, Hartree-Fock method, andmolecular dynamics in Li-S batteries cathode, electrolyte, and anode. At last, a conclusion is draw and a perspective is present for first-principles calculation.
first-principles calculation; lithium-sulfur batteries; sulfur cathode; electrolyte; lithium anode
10.12028/j.issn.2095-4239.2017.0031
TM 911
A
2095-4239(2017)03-500-22
2017-03-27;
2017-04-14。
国家重点研发计划(2016YFA0202500),国家重点基础研究发展计划(2015CB932500),国家自然科学基金项目(21676160)。
陈翔(1994—),男,博士研究生,主要研究方向为能源材料理论计算,E-mail:chenxiang16@mails.tsinghua.edu.cn;
张强,研究员,主要研究方向为能源材料,尤其是金属锂、锂硫电池和电催化剂,E-mail:zhang-qiang@mails.tsinghua.edu.cn。
