不伴感觉神经受累的肯尼迪病5例临床特征分析
2017-10-12周晓萌刘亚玲冉冀娜王莹
周晓萌 刘亚玲 冉冀娜 王莹○☆
不伴感觉神经受累的肯尼迪病5例临床特征分析
周晓萌*刘亚玲*冉冀娜*王莹*○☆
目的分析不伴有感觉异常肯尼迪病的临床特征、血清学检查、电生理检查等,以指导临床诊断降低误诊率。方法收集经基因明确诊断的肯尼迪病5例,详细询问其病史,进行全面的体格检查包括详尽的神经系统查体,收集并分析其实验室检查指标、电生理检查特点,及基因测定AR基因1号外显子CAG重复序列。结果5例患者均无明显阳性家族史,均为男性,平均起病年龄(39.8±7.2)岁,从发病到确诊平均病程(9.0±5.2)年,3例患者起病部位为双下肢近端无力,1例患者为口周及颊部"肉跳"感,1例患者为男性乳腺发育;最显著的临床表现为舌肌萎缩、舌肌纤颤、四肢近端肌肉无力;5例患者均无临床及电生理测定的感觉异常。结论肯尼迪病是一种累及下运动神经元的神经变性疾病,感觉不受累的患者也应考虑肯尼迪病,确诊依赖基因测定。
肯尼迪病 AR突变蛋白 多系统病变
肯尼迪病(Kennedy disease,KD)又称脊髓延髓肌萎缩症 (spinal and bulbar muscular atrophy,SBMA),是一种X连锁隐性遗传的下运动神经元变性疾病。其临床表现为缓慢进展的肢体及延髓部肌肉萎缩、无力、肌束震颤[1],与肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)或运动神经元病(motor neuron disease,MND)极易混淆,给患者及家庭造成极大的恐惧。因此,对肯尼迪病的正确诊断尤为重要,本文就我科近期经基因确诊为肯尼迪病5例报吿如下。
1 对象与方法
1.1 研究对象患者来源于2012~2017年由河北医科大学第二医院神经内科确诊的肯尼迪病5例,5例患者均无阳性家族史。
1.2.1 临床资料收集 详细询问其发病年龄,首发症状,尤其注重询问其肌肉无力及肌肉萎缩情况,肌束震颤分布等,询问其家族史情况,尤其其家族中男性成员的患病情况;详细询问生育史及性功能;并进行神经系统及全身体格检查,包括肌肉萎缩分布、四肢肌力、病理征、观察有无男性乳腺发育;完善血清学检查,尤其是肌酸激酶测定,及其他代谢、内分泌相关指标测定;收集完整针极肌电图及运动、感觉神经传导速度测定,观察患者肌电图测定相关的急性失神经表现及慢性神经再生情况,及有无感觉神经受累。同时收集患者基因测定结果。
1.2.2 基因测定结果 应用PCR技术测定患者AR基因1号外显子N端CAG重复序列(图1)
2 结果
2.1 临床表现及体格检查见表1,一般特点:患者均为男性;发病年龄:25~61 岁,平均(39.8±7.2)岁;确诊时发病时间为 2~28 年,平均(9.0±5.2)年;起病方式:3例双下肢近端无力,1例口周及颊部“肉跳”,1例为男性乳腺发育;临床表现:肌肉无力表现为面部肌肉无力及四肢近端无力,其中5例均为近端肌无力,2例出现中枢性面瘫,3例有构音障碍,1例表现为咀嚼肌无力;肌束震颤较广泛其中4例有面部明显肌肉肌束震颤,1例全身肌束震颤;肌肉萎缩多累及舌肌、四肢肌肉,其中5例均有明显舌肌萎缩(图1),1例左侧腓肠肌萎缩、双手大鱼际肌饱满度减低、双手第一骨间肌萎缩,1例双侧肩胛肌萎缩;四肢腱反射多减弱或正常其中 4 例腱反射(+),1 例腱反射(++);病理反射均未引出;感觉系统查体均未见明显异常;雄激素不敏感表现可见男性乳房发育、性能力下降、不育,其中4例均有男性乳房发育(图2),1例有性功能减低伴不育。
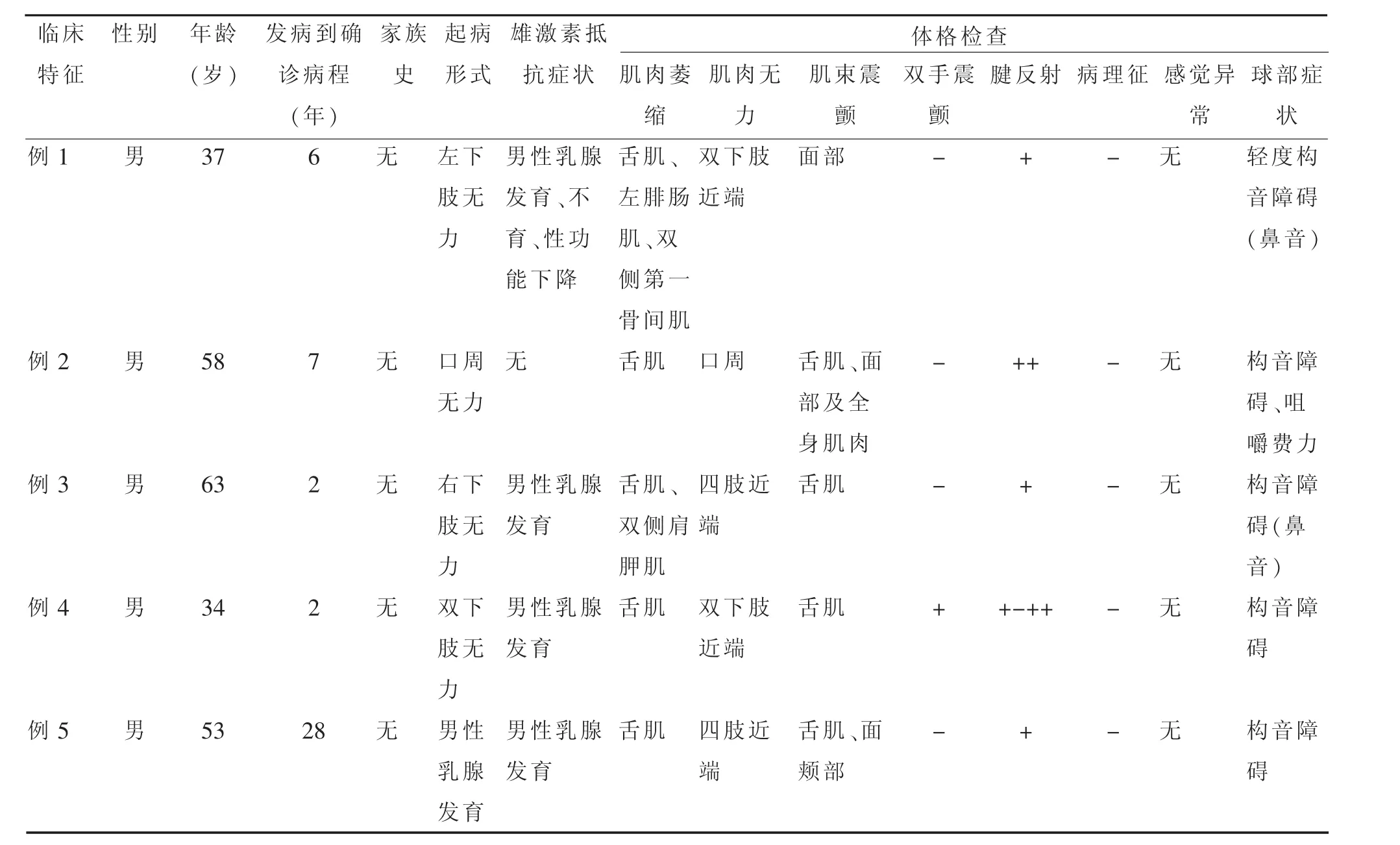
表1 5例肯尼迪病患者临床特点分析
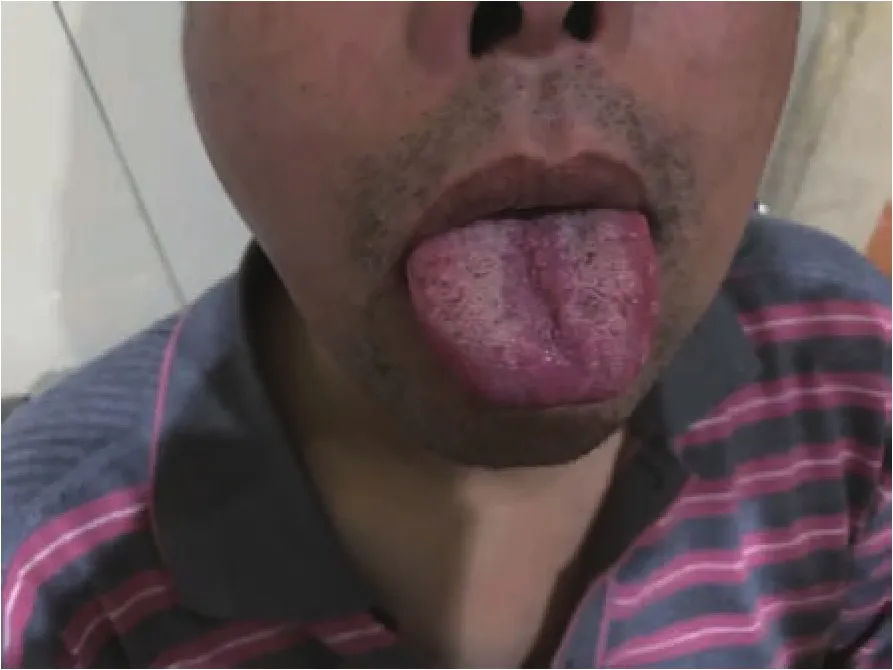
图1 患者例5左侧舌肌萎缩
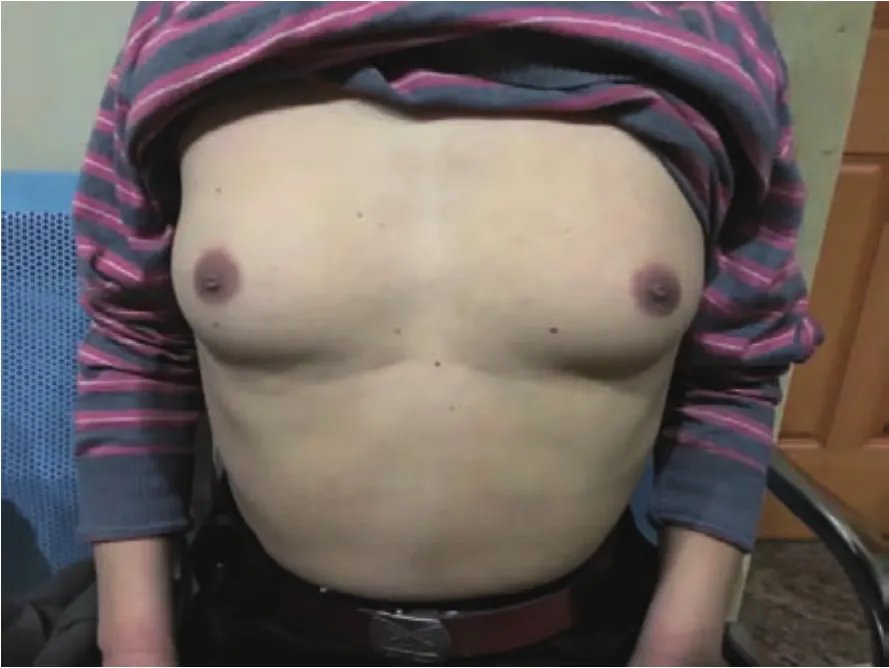
图2 患者例5男性乳腺发育
2.2 电生理检查:见表2常规肌电图检查示被检肌(颈、胸、腰、球段)呈广泛神经源性损害;感觉神经传导速度 (sensory nerve conduction velocity,SCV)检查均未见明显异常;运动神经传导速度(motor nerve conduction velocity,MCV):1 例 正常,2例右正中神经、右腓总神经复合肌肉动作电位波幅减低,2例腓总神经复合肌肉动作电位波幅减低。
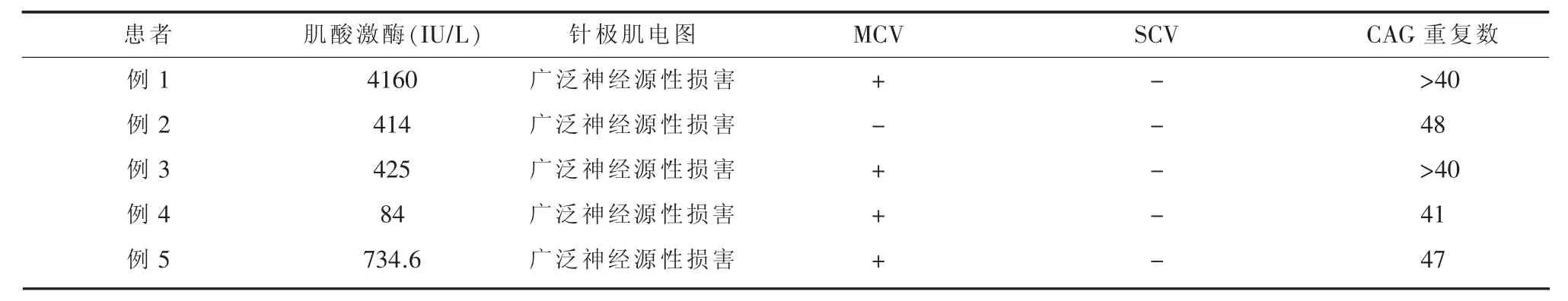
表2 5例肯尼迪病患者血清学及电生理检查特点
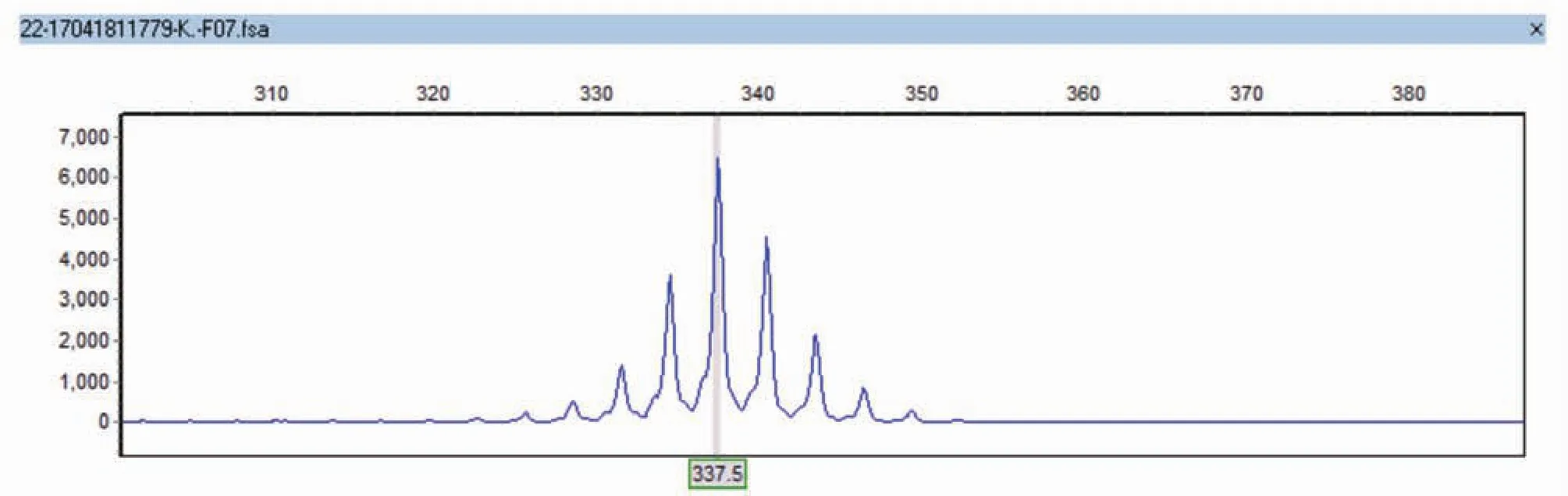
图3 患者例5 AR基因1号外显子CAG重复序列
2.3 血清学检查:见表2肌酸激酶检查为轻中度升高;内分泌系统检查提示存在葡萄糖及脂肪代谢异常,其中1例合并高脂血症,1例伴有脂肪肝,1例合并2型糖尿病,1例合并高甘油三酯血症、高尿酸血症,转氨酶轻度升高。
2.4 基因检测:AR基因1号外显子N端CAG重复序列均大于40(图1)。
3 讨论
肯尼迪病(Kennedy disease KD)由美国医生KENNEDY等[2]于1968年首次报告。1999年,LA SPADA等[1]首次证实致病基因位于Xql1-12的雄性激素受体(androgen receptor,AR)基因1号外显子,因其N端CAG重复序列异常扩增,导致雄激素受体蛋白内的poly Q区域扩增而致病。当CAG扩增数目达到38~62时会引起肯尼迪病[3]。
肯尼迪病在不同种族间的发病率不同,国外一项20例肯尼迪病研究称肯尼迪病发病率为1/50000男性,仅男性患肯尼迪病,而女性患者即使是突变的纯合子也仅表现为亚临床症状[4],台湾一项报道称肯尼迪病发病率为1/40000男性[5];发病年龄较早,多在青中年出现临床症状,病程较长,10年存活率为82%[6-8]。本研究5例患者与既往文献报告相一致。
肯尼迪病病因学研究较多主要集中于AR基因CAG序列的异常扩增翻译后的雄激素受体(AR)蛋白的毒性作用,突变蛋白广泛存在于人体各器官中,分布于脑干运动神经元、脊髓前角细胞[2],引起支配肌肉的萎缩及无力;该蛋白同时还存在于脊髓背根神经节细胞而出现较轻微的肢体远端的感觉减退[9];雄激素受体蛋白亦可表达于骨骼肌细胞,对骨骼肌细胞存在直接毒性作用,出现血清肌酸激酶的升高[10]。本研究5例患者均存在不同程度的肌肉萎缩、肌肉无力、肌酸激酶轻中度上高,例4患者肌酸激酶正常,但均不存在主观及电生理测定的感觉异常,与既往文献报道有差异。最新报道称雄激素受体蛋白在人体肾脏、肾上腺和阴囊皮肤细胞中均有表达,活检表明阴囊皮肤上皮细胞中AR蛋白的聚集程度与脑干运动神经元中聚集程度相吻合,且与CAG重复次数相关,与运动功能呈负相关,因此提议用检测阴囊皮肤上皮细胞内的多聚谷氨酰胺作为监测肯尼迪病病程的生物标记物[11],这一观点仍有待于进一步研究证实。
肯尼迪病由于失去正常AR蛋白的功能而出现雄激素不敏感的表现如男性乳房女性化、少精子症、不育、勃起功能障碍等[6],由于AR蛋白分布广泛,因此肯尼迪病患者会出现多个器官受累的表现[12],累及内分泌系统出现脂肪、葡萄糖代谢异常等。本研究4例患者均有雄激素不敏感症状;内分泌系统检查提示存在葡萄糖及脂肪代谢异常,其中例4合并2型糖尿病,例1伴有脂肪肝同时合并高甘油三酯血症、高尿酸血症,转氨酶轻度升高,例3伴有脂肪肝。
尽管肯尼迪病的临床特征较多,但是明确首发症状对肯尼迪病的诊断至关重要。首发症状国内报道多以肢体无力起病,有以下肢力弱起病,四肢力弱起病,上肢力弱起病[8];而国外文献报告首发症状为男性乳房发育,肌肉疼痛,过早肌肉疲劳感,而肢体无力并不是典型的早期症状,如果出现通常为肢体末梢无力,面部肌肉震颤较明显[7],国内外均有文献报道姿势性肢体震颤可能为肯尼迪病临床症状出现前最早的表现。本研究中3例患者首发症状为下肢力弱起病,1例患者以口周无力及面颊部“肉跳”感为首发症状,1例患者追溯首发症状为男性乳腺发育;3例患者均出现明显的面部肌束震颤;1例患者出现明显双手震颤,本研究中患者起病方式与国内研究报道相一致,与国外文献报道略有差异,可能与种族差异有关。
肯尼迪病的早期诊断有赖于神经电生理检查,鲁明等[13]总结的12例肯尼迪病患者肌电图及神经电图的特点:EMG呈神经源性改变,感觉神经动作电位波幅降低,感觉神经传导速度减慢;国外34例肯尼迪病患者研究报告50%患者可出现感觉神经系统受累的表现[7]。本研究5例患者常规肌电图检查与既往文献报告一致,而神经电图测定均无感觉神经传导异常。
肯尼迪病目前无有效的治疗方法,而亮丙瑞林、ASC-J9等药物有望成为治疗肯尼迪病的新型药物[14],对症治疗对患者预后有一定益处。由于肯尼迪病临床特征与肌萎缩侧索硬化极易混淆,国外一项研究报告2%的肌萎缩侧索硬化患者最终确诊为肯尼迪病[15]。因两种疾病不同的病因学及预后,临床工作中遇到疑似运动神经元病的男性患者应常规检查男性乳房发育情况,询问雄激素受体不敏感的症状,最终明确鉴别有赖于基因检测,随着基因检测技术的推进,肯尼迪病的误诊率也在逐渐降低,对病人及家庭而言均大有裨益。
[1]LA SPADA AR,WILSON EM,LU DB,et al.Androgen receptor mutation in X-linked spinal and bulbar muscular atrophy[J].Nature,1991,352:77-79.
[2]KENNEDY WR,ALTER M,SUNG JH.Progressive proximal spinal and bulbar muscular atrophy of late onset.A sex-linked recessive trait[J].Neurology,1968,18(7):671-680.
[3]ATSUTA N,WATANABE H,ITO M,et al.Natural history of spinal and bulbar muscular atrophy(SBMA):a sudy of 223 Japanese patients[J].Brain,2006,129(6):1446-1455.
[4]ELISA DI ROSA,GIANNI SORARU,JOHANN ROLAND KLEINBUB.Theory of mind,empathy and neuropsychological functioning in X-linked Spinal and Bulbar Muscular Atrophy:a controlled study of 20 patients[J].Neurol,2015,262(2):394-401.
[5]MU-HUI FU,MIN-YU LAN,JIA-SHOU LIU,et al.Kennedy Disease Mimics Amyotrophic Lateral Sclerosis:A Case Report[J].Acta Neurol Taiwan,2008,17(7):99-103.
[6]CHAHIN N,KLEIN C,MANDREKAR J,et a1.Natural history of spinal-bulbar muscular atrophy.Neurology,2008,70(21):1967-1971.
[7]ANNE D,SPERFELD MD,JOCHEM KARITZKY MD,et al.X-linked Bulbospinal Neuronopathy,Kenndy Disease [J].Arch Neurol,2002,59(12):1921-1926.
[8]崔丽英,刘明生,谢曼青,等.肯尼迪病基因诊断及临床特点[J].中华医学杂志,2010,90(35):2498-2500.
[9]SOBUE G,MATSUOKA Y,MUKAI E,TAKAYANAGI T,SOBUE I.Pathology of myelinated fibers in cervical and lumbar ventral spinal root in amyotrophic lateral sclerosis[J].Neurol Sci,1981,50(3):413-421.
[10]YU Z,DADGER N,ALBERTELLI M,et al.Androgendependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model[J].Clin Invest,2006,116(10):2663-2672.
[11]KATSUNO M,BANNO H,SUZUKI K,et al.Molecular genetics and biomarkers of polyglutamine diseases[J].Curr Mol Med,2008,8(3):221-234.
[12]POLETTI A.The polyglutamine tract of androgen receptor:from functions to dysfunctions in motor neurons [J].Front Neuroendocrinol,2004,26(1):1-26.
[13]鲁明,张俊,郑菊阳,等.12例肯尼迪病患者肌电图和神经电图特点[J].中国神经免疫学和神经病学杂志,2008,15(3):187-189.
[14]何炳接,何若洁,石磊.肯尼迪病临床特征与CAG重复序列数目关系分析 [J].中国神经精神疾病杂志,2015,41(9):547-551.
[15]PAPAROUNAS K,GOTSI A,SYRROU M,et al.Kennedy disease:avoiding misdiagnosis[J].Arch Neurol,2003,60(6):893-894.
Clinical features of Kennedy's disease without sensory nerve involvement:a report of 5 cases.
ZHOU Xiaomeng,LIU Yaling,RAN Jina,WANG Ying.Department of Pathology,the Second Hospital of Hebei Medical University,Shijiazhuang 050000.Tel:0311-66003942.
ObjectiveTo analyze the clinical feature,serum examination,EMG of Kennedy'Disease to reduce misdiagnosis of Kennedy's Disease.MethodsFive casesof Kennedy's disease were confirmed by genetic test.The clinical data was analyzed including clinical features,laboratory findings,EMG characteristics and determination of AR gene exon 1 CAG repeat sequence.ResultsThese cases were male without an obvious positive family history.The average age of onset was 39.8 ±7.2 years old and the average duration from onset to diagnosis was 9 ±5.2 years.Onset symptoms included Lower limbs weakness in 3 cases,facial fasciculationin 1 cases and gynecomastia in 1 case.The most prominent clinical manifestations were tongue muscle atrophy,tongue muscle fibrillation and proximal limb muscle weakness.In addition,these 5 cases did not have clinical manifestation of sensation loss nor EMG evidence of abnormal sensation.ConclusionKennedy's disease is a neurodegenerative disease characterized by lower motor neuron damage.The clinical features of these 5 cases are approximately the same as those reported in previous literatures.Although the patients have been reported to have abnormal sensation,the present study indicates that some patients with Kennedy's disease may not present with abnormal sensation and that the diagnosis of Kennedy's disease depends on the genetic test.
Kennedy'Disease AR mutation protein Multisystem lesion
R744.8
A
2017-03-02)
(责任编辑:李立)
10.3969/j.issn.1002-0152.2017.08.008
* 河北医科大学第二医院(石家庄 050000)
○☆通信作者(E-mail:lyldoctor@163.com)
