Letters to the Editor
2017-10-09
Letters to the Editor
Polymorphic multiple hepatocellular adenoma including a non-steatotic HNF1α-inactivated variant
To the Editor:
Hepatocellular adenomas (HCAs) consist of benign liver tumors favored by the use of oral contraceptives, which preferentially occur in women.[1,2]ey expose to the risk of hemorrhage (20% of cases) and more rarely, to the risk of malignant transformation (4%-10% of cases).[3,4]Multiple HCAs, which are defined by the presence of 10 or more HCAs, were first described in 1985 and were initially considered having a worse prognosis than single HCA.[5]However, recent reports have suggested that the risk of complications in patients with multiple HCAs was not related to the number of lesions, but mainly to three other factors: i) tumor size >5 cm for the risk of both hemorrhage and malignant transformation;[3]ii) male gender and iii) β-catenin-mutation for the risk of malignant transformation.[6]Progresses made in terms of genotypic knowledge of HCA have allowed to identify different subtypes of HCA associated with more specific risks of complication.[7]A recent report has suggested that some patients could present with mixed subtypes of HCA.[8]Herein, we report the case of a female patient presenting with polymorphic multiple HCA of three different subtypes, including a very unusual non-steatotic variant of hepatocyte nuclear factor 1 homeobox α (HNF1α)-inactivated HCA mimicking malignant transformation,which was also complicated with repeated episodes of hemorrhage and was finally managed with liver transplantation.
A 34-year-old female patient was admitted at the emergency department of our institution in September 2011 for acute right upper quadrant abdominal pain. Her past medical history included mild overweight (BMI=26.1 kg/m2), epilepsy, appendectomy and the use of oral contraceptive for 10 years, which had been stopped for 5 years. Blood tests revealed elevated levels of C-reactive protein (CRP) (179 mg/L), liver enzymes (serum aspartate aminotransferase 536 UI/L, gamma-glutamyl transferase 119 UI/L, alkaline phosphatase 267 UI/L) were high, while the red blood cell count and alpha-fetoprotein levels were normal. Magnetic resonance imaging (MRI) showed an 8-cm large hypervascular tumor located in liver segment 7 complicated with a 7-cm large intrahepatic hematoma, and multiple other hypervascular nodules suggesting the co-existence of steatotic and inflammatory HCAs (Fig. 1).

Fig. 1. Imaging and gross appearance of polymorphic multiple hepatocellular adenomas. Transverse T1-weighted magnetic resonance imaging with intravenous Gadolinum injection showed a large hepatocellular adenoma (arrow) with a subcapsular hematoma (arrow heads) (A). The gross appearance after fixation in formalin showed an 8-cm large soft, rounded, well-demarcated nodule, with smooth borders (arrow heads), with an area showing hemorrhage (arrow) (B). Some of the lesions were consistent with steatotic hepatocellular adenoma, showing an iso-signal on T1-weighted sequence (C), with important drop out signal on chemical shisequence due to its fatty content (D), and a slight arterial enhancement on T1 fat sat sequence aer Gadolinium injection (E), which vanished on late phase (F). Some other lesions were inflammatory, showing a slightly hyper intense “in phase”signal on T1-weighted sequence (G) without drop out signal on chemical shisequences (H). It showed strong arterial enhancement (I) and heterogeneous signal on late phase (J).
Close monitoring was advocated until hematoma disappearance before considering a more specific management. Two months later, the patient was readmitted for recurrent abdominal pain. Liver MRI showed intra-tumoral bleeding in another nodule located in segment 6,while that same lesion was 30-mm large and was uncomplicated on the initial CT performed two months earlier.By contrast, the hematoma in segment 7 decreased in size. Several tests were performed and precluded underlying metabolic disorders that could favor multiple HCA,such as glycogen storage disease, hormonal disorders and type 3 maturity onset of diabetes of the young. Due to the repeated episodes of hemorrhage, hepatectomy was undertaken.
In January 2012, the patient underwent a right hemihepatectomy associated with tumorectomy of a single 30-mm nodule located in the lelobe. Surgical specimen revealed 8 nodules grossly, of which two (30 and 80 mm in size) had hemorrhagic infarction, whereas some twenty additional hepatocellular micronodules ranging from 1 to 8 mm were identified on histology (Table). Morphology and immunostains with liver fatty acid binding protein (LFABP), CRP, serum amyloid A (SAA), glutamine synthetase (GS), and β-catenin allowed us to easily classify some of them as HCAs and focal nodular hyperplasia(FNH).ose typical HCAs were classified into inflammatory (SAA positive, CRP positive, LFABP positive) or steatotic HNF1α-inactivated HCA (SAA negative, CRP negative, LFABP negative).[9]In addition to those benign hepatocellular nodules, a second group of 11 nodules displayed worrisome features, such as a pseudoglandular pattern and some cellular monotony, with an SAA negative,CRP negative and LFABP positive phenotype (Fig. 2). Of these 11 nodules, 8 were considered atypical or borderline between HCA and hepatocellular carcinoma: all these atypical nodules lacked steatosis, inflammatory infiltrates,ductular reaction and reticulin loss. No vascular invasion was seen and only one small 7-mm nodule displayed GSstaining. On the basis of the International Consensus Group for Hepatocellular Neoplasia,[10]the remaining three nodules were finally considered grade 1 hepatocellular carcinoma due to a nodule-in-nodule pattern within a large inflammatory HCA, or because of extensive pseudoacinar changes associated with mild atypia and some increase in cell density.e initial diagnosis performed in January 2012 was that of mixed adenomatosis with atypical unclassified HCAs (n=8; size range: 2-10 mm) and adenoma with malignant transformation into grade 1 hepatocellular carcinoma (one 35-mm lesion and one lesion consisting of 3 incipient foci within an 80 mm inflammatory HCA).
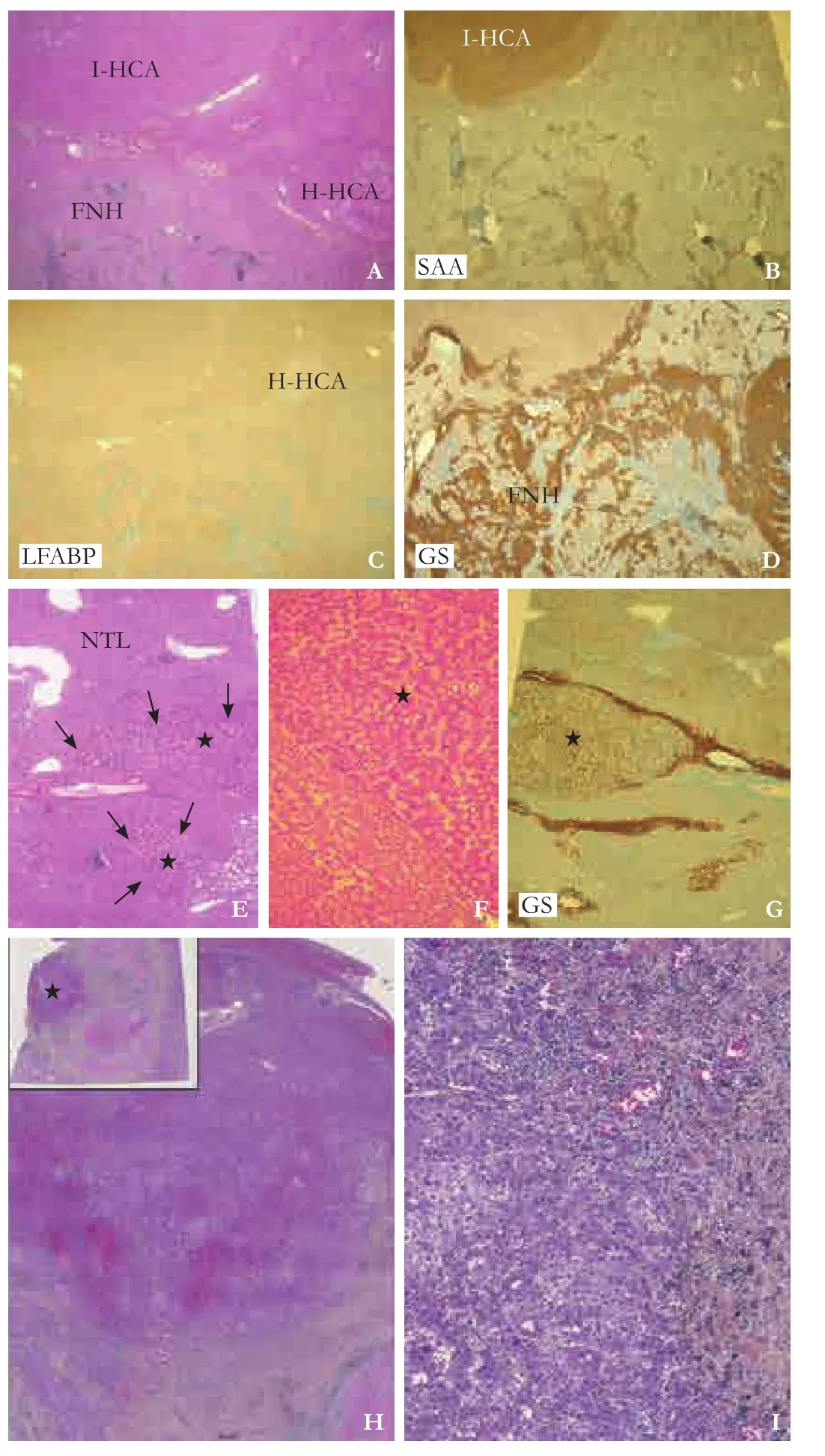
Fig. 2. Surgical specimen aer right hemihepatectomy. Pathology of the specimen showed the coexistence of inflammatory hepatocellular adenoma (I-HCA), HNF1α-inactivated hepatocellular adenoma(H-HCA) and focal nodular hyperplasia (FNH) (A, HE stain, ×20),with positive serum amyloid A (SAA) in I-HCA (B, HE stain, ×20),loss of expression of liver fatty acid binding protein (LFABP) in H-HCA(C, HE stain, ×20), and reactivity for glutamine synthetase (GS) in FNH (D, immunohistochemistry, ×20). Some lesions presented with a pattern of nodule in nodule mimicking hepatocellular carcinoma due to the presence of 2 microscopic foci of non-steatotic HNF1α-inactivated adenoma (arrows and stars, E, HE stain, ×40) within a large 8-cm inflammatory adenoma, with numerous pseudoglandular structures, monotonous hepatocytes and mild increase in nuclear density (F, HE stain, ×100), and no expression of GS (G, immunohistochemistry, ×20). Another lesion was defined as a borderline nodule due to the presence of necrotico-hemorrhage within a 4-cm nodule with peripheral residual tumor (star, H, HE stain, ×20), numerous acinar structures, cellular monotony, increase in nuclear density and mild nuclear atypia (I, HE stain, ×200). NTL: non-tumorous liver.
Table. Summary of histopathological findings on specimen aer right hemihepatectomy and liver transplantation

Table. Summary of histopathological findings on specimen aer right hemihepatectomy and liver transplantation
HCA: hepatocellular adenoma.
Right hemihepatectomy Explant Histopathological type Number of nodules Size range(mm)HNF1α-inactivated HCA Size range(mm)Number of nodules 3 1-8036 1-20 Inflammatory HCA3 5-30 3 5-15 Atypical HCA8 7-40 1 5 Hepatocellular carcinoma 320-35 1 2 Focal nodular hyperplasia 2 8-50 130 Incipiens benign hepatocellular nodule 8 1-9 0-Necrotic nodule230-40 312-40
During follow-up, 2 nodules of less than 30 mm located within the remnant lelobe increased in size (26 to 34 mm and 19 to 30 mm, respectively). Because of rapid size progression and previous pathological findings suggesting multiple HCC transformations, the patient was considered for orthotopic liver transplantation, and aer board approval she was listed and underwent orthotopic liver transplantation one year aer initial partial hepatectomy. Specimen of the explant showed a total of 36 steatotic HCAs with loss of LFABP expression associated with three inflammatory HCAs, including one with malignant transformation, and one atypical HCA (Table).Aer a follow-up of 40 months, the patient was alive without recurrence.
Additional genetic study of one “malignant” and 4 atypical nodules was recently conducted and showed absence of mutation of IL6ST, GNAS, β-catenin, and TERT promoter genes, and it also demonstrated somatic inactivating mutations of HNF1α in this subgroup, permitting the revised diagnosis of a non-steatotic variant of HNF1α-inactivated HCA.erefore, the definitive diagnosis is that of mixed adenomatosis with coexistence of typical inflammatory HCAs, HNF1α-inactivated HCAs, and uncommon non-steatotic variants of HNF1α-inactivated HCA.
Tumor bleeding is mainly expected to occur in patients with HCA greater than 5 cm[3]and episodes of clinically relevant hemorrhage have been seldom reported for tumors smaller than 5 cm.[11-14]However, in the present case, 2 consecutive episodes of tumor hemorrhage occurred in less than 4 months, and concerned 2 different HCAs, of which 1 was less than 3 cm large.ese findings suggest that the risk of hemorrhage, albeit low, does exist for HCA of less than 5 cm, and therefore,close monitoring must be considered even for patients with small HCA.
In most cases, all HCAs in a given patient with multiple HCA are considered the same subtype. However,Castain et al[8]reported in 2014 the first well-documented series of polymorphic HCAs of 9 patients with coexisting HNF1α-inactivated and inflammatory HCAs.e present observation consists of one additional rare case of mixed polymorphic adenomatosis, and to our knowledge, this was the first showing the coexistence of three phenotypically distinct HCAs, including two typical subtypes (inflammatory and HNF1α-inactivated) and one very unusual variant of HNF1α-inactivated HCA with lack of steatosis.
All HCAs were considered for surgery more than ten years ago - some of them being even considered for liver transplantation[17]-only selected cases are nowadays considered for hepatectomy,[3]and there are almost no more indications lefor transplantation, except for patients with glycogen storage type 1 disease,[18]and for patients with a high-risk of hemorrhage or malignant transformation, who are not amenable to surgical resection.[19,20]In the present case, liver transplantation was undertaken because of the initial diagnosis of multiple malignant transformation aer partial hepatectomy, and the rapid increase of size, suggesting a potential risk for hemorrhage and further malignant transformation.
Kayvan Mohkam, Benjamin Darnis, Jean-Baptiste Cazauran, Agnès Rode, Anne-Frédérique Manichon, Christian Ducerf, Brigitte Bancel and Jean-Yves Mabrut
Department of General Surgery and Liver Transplantation (Mohkam K, Darnis B, Cazauran JB, Ducerf C and Mabrut JY), Department of Radiology (Rode A and Manichon AF), Department of Surgical Pathology (Bancel B),Hospices Civils de Lyon, Croix-Rousse University Hospital,Lyon, France; Equipe Mixte de Recherche 3738, Ecole Doctorale EDISS 205, Université Lyon 1, France (Mohkam K,Darnis B and Mabrut JY)
Acknowledgments:We thank Salim Mezoughi and Hassan G Demian for their contribution to the manuscript.
Contributors:DC and MJY proposed the study, interpreted the data, and critically revised the manuscript. MK, DB and CJB collected data, and draed the manuscript. RA and MAF performed the imaging analysis and revised the manuscript. BB analyzed and classified the data and wrote the manuscript. MK is the guarantor.
Funding:None.
Ethical approval:Informed consent was obtained from the patient and the reporting of this case was approved by the institutional scientific board.
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Rooks JB, Ory HW, Ishak KG, Strauss LT, Greenspan JR, Hill AP, et al. Epidemiology of hepatocellular adenoma.e role of oral contraceptive use. JAMA 1979;242:644-648.
2 Rebouissou S, Bioulac-Sage P, Zucman-Rossi J. Molecular pathogenesis offocal nodular hyperplasia and hepatocellular adenoma. J Hepatol 2008;48:163-170.
3 Dokmak S, Paradis V, Vilgrain V, Sauvanet A, Farges O, Valla D,et al. A single-center surgical experience of 122 patients with single and multiple hepatocellular adenomas. Gastroenterology 2009;137:1698-1705.
4 Farges O, Ferreira N, Dokmak S, Belghiti J, Bedossa P, Paradis V,et al. Changing trends in malignant transformation of hepatocellular adenoma. Gut 2011;60:85-89.
5 Flejou JF, Barge J, Menu Y, Degott C, Bismuth H, Potet F, et al.Liver adenomatosis. An entity distinct from liver adenoma?Gastroenterology 1985;89:1132-1138.
6 Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C,Rebouissou S, et al. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology 2006;43:515-524.
7 Bioulac-Sage P, Laumonier H, Couchy G, Le Bail B, Sa Cunha A, Rullier A, et al. Hepatocellular adenoma management and phenotypic classification: the Bordeaux experience. Hepatology 2009;50:481-489.
8 Castain C, Sempoux C, Brunt EM, Causse O, Heitzmann A,Hernandez-Prera JC, et al. Coexistence of inflammatory hepatocellular adenomas with HNF1α-inactivated adenomas: is there an association? Histopathology 2014;64:890-895.
9 Bioulac-Sage P, Cubel G, Balabaud C, Zucman-Rossi J. Revisiting the pathology of resected benign hepatocellular nodules using new immunohistochemical markers. Semin Liver Dis 2011;31:91-103.
10 International Consensus Group for Hepatocellular Neoplasiae International Consensus Group for Hepatocellular Neoplasia. Pathologic diagnosis of early hepatocellular carcinoma:a report of the international consensus group for hepatocellular neoplasia. Hepatology 2009;49:658-664.
11 Deneve JL, Pawlik TM, Cunningham S, Clary B, Reddy S,Scoggins CR, et al. Liver cell adenoma: a multicenter analysis of risk factors for rupture and malignancy. Ann Surg Oncol 2009;16:640-648.
12 Cho SW, Marsh JW, Steel J, Holloway SE, Heckman JT, Ochoa ER, et al. Surgical management of hepatocellular adenoma:take it or leave it? Ann Surg Oncol 2008;15:2795-2803.
13 van der Windt DJ, Kok NF, Hussain SM, Zondervan PE,Alwayn IP, de Man RA, et al. Case-orientated approach to the management of hepatocellular adenoma. Br J Surg 2006;93:1495-1502.
14 Ribeiro A, Burgart LJ, Nagorney DM, Gores GJ. Management of liver adenomatosis: results with a conservative surgical approach. Liver Transpl Surg 1998;4:388-398.
15 Sempoux C, Paradis V, Komuta M, Wee A, Calderaro J, Balabaud C, et al. Hepatocellular nodules expressing markers of hepatocellular adenomas in Budd-Chiari syndrome and other rare hepatic vascular disorders. J Hepatol 2015;63:1173-1180.
16 Arrivé L, Zucman-Rossi J, Balladur P, Wendum D. Hepatocellular adenoma with malignant transformation in a patient with neonatal portal vein thrombosis. Hepatology 2016;64:675-677.17 Chiche L, Dao T, Salamé E, Galais MP, Bouvard N, Schmutz G,et al. Liver adenomatosis: reappraisal, diagnosis, and surgical management: eight new cases and review of the literature. Ann Surg 2000;231:74-81.
18 Lerut JP, Ciccarelli O, Sempoux C, Danse E, deFlandre J,Horsmans Y, et al. Glycogenosis storage type I diseases and evolutive adenomatosis: an indication for liver transplantation.Transpl Int 2003;16:879-884.
19 Di Sandro S, Slim AO, Lauterio A, Giacomoni A, Mangoni I,Aseni P, et al. Liver adenomatosis: a rare indication for living donor liver transplantation. Transplant Proc 2009;41:1375-1377.
20 Chiche L, David A, Adam R, Oliverius MM, Klempnauer J,Vibert E, et al. Liver transplantation for adenomatosis: European experience. Liver Transpl 2016;22:516-526.
Published online September 4, 2017.
Gastrointestinal tract post-transplant lymphoproliferative disorder aer liver transplantation
To the Editor:
Post-transplant lymphoproliferative disorder (PTLD) is a rare and potentially fatal complication occurring aer all types of solid organ transplantation.[1]PTLD accounts for 20% of allde novopost-transplant tumors.[2,3]e most important risk factors for PTLD are prolonged intense immunosuppression and Epstein-Barr virus (EBV)infection.[4]e gastrointestinal (GI) tract is the most frequently involved site (GI-PTLD), the liver allograitself can also be involved.[5]As clinical manifestations of PTLD may vary, early diagnosis of PTLD is oen difficult.[6,7]
Pathology together with appropriate immunohistochemical detection of EBV infection is the gold standard for diagnosing PTLD.e more frequent localization in the GI tract may be explained by the presence of the rich gut-associated lymphoid tissue as well as antigen-presenting cells.is is in contrast to the fact that the normal liver does not contain lymphoid follicles composed of B or T lymphocytes.[8,9]
Based on our single-center experience, clinicopathological features and the treatment options of GI-PTLD in the context of liver transplantation are discussed.e characteristics of the four patients presenting GI-PTLD are displayed in Table. Pathology revealed in all four patients diffuse infiltrate of large atypical lymphocytes with irregular nuclei and prominent nucleoli. Immunohistochemistry staining showed the large monoclonal lymphocytes, which were strongly and uniformly positive for CD20, CD79a, and negative for CD3, CD5, CD10 and Bcl-6. Ki-67 index was high. EBV encoded RNA (EBER)was demonstrated in two patients byin situhybridization(Fig. 1).
Case 1: A 13-year-old girl who underwent OLT three years earlier because of congenital liver fibrosis was readmitted to the hospital due to bloody stool and fever.Colonoscopy showed multiple, 0.8 to 1.0 cm large, ulcersin the colon; capsule endoscopy further revealed numerous irregular, bleeding, jejunal and ileal ulcers, corresponding at biopsy to DLBCL in the presence of EBV infection (EBV-DNA virus load: 2.28×104copies/mL);bone marrow was normal (Fig. 1). Treatment consisted of discontinuation of tacrolimus and introduction of low-dose steroid followed by four cycles of intravenous rituximab, these treatment resulted in complete remission of the disease up till 27.5 months.

Table. Clinical and pathologic features of GI-PTLD cases
Case 2: A 51-year-old male transplanted for eleven years because of HBV-related cirrhosis was readmitted because offever, abdominal pain and anemia. Endoscopy revealed multiple gastric and duodenal ulcers, with a diameter up to 3.5 cm corresponding again to DLBCL(Fig. 2A). Markers for EBV infection were negative as the bone marrow. Six cycles of CHOP chemotherapy (cyclophosphamide, hydroxydaunorubicin, oncovin and prednisone) allowed to obtain complete remission confirmed by successive endoscopy at 6 and 36-month follow-up(Fig. 2 B, C). Due to the fact that he had a history of several acute cellular rejections before, immunosuppression was not reduced. During the chemotherapy, he developed pulmonary tuberculosis necessitating therapy and the CHOP dose was reduced. Unfortunately, he needed a re-transplantation 14-month aer diagnosis of DLBCL because of drug induced acute liver failure. He is now alive and disease free at 43 months.
Case 3: A 61-year-old male, transplanted twelve years before because of HBV-related cirrhosis was readmitted due to recurrent abdominal pain. Abdominal CT-scan revealed a thickened terminal ileum wall in the presence of multiple intraluminal papillary lesions. Colonoscopy confirmed a stiffened terminal ileum wall in the presence of an obstructive tumor, suggestive of PTLD.e pathology of the right hemicolectomy specimen revealed a 7×6 cm ulcerated tumor extending into the ileum, appendix and adjacent serosa corresponding to an EBV-negative DLBCL; bone marrow was negative for PTLD. Chemotherapy consisting of 8 cycles of intravenous rituximab and 3 cycles of R-CHOP followed by a right hemicolectomy, resulting in complete disease remission. He was disease free aer 46 months offollow-up.
Case 4: A 60-year-old male, transplanted thirteen years before due to HBV-related cirrhosis, was readmitted because of abdominal pain and bloody stool.Abdominal CT-scan identified a rectal mass involving perirectal fat as well as enlarged bilateral inguinal and retroperitoneal lymph nodes. Rectoscopy identified an ulcerated, bleeding rectal mass. Radical proctocolectomy with terminal ileostomy was performed and followed by chemotherapy consisting of 3 cycles of R-CHOP, 2 cycles R-COMP (rituximab-cyclophosphamide, vindesine, mitomycine and prednisone) and 3 cycles of COMP, 1 cycle each of COP-E (cyclophosphamide, vindesine, prednisone, etoposide) and MOP (mitomycine, vindesine, prednisone).e pathology report confirmed the diagnosis of EBV-positive DLBCL; bone marrow was normal. He was disease free at 139 months offollow-up.

Fig. 1.Histopathology of PTLD in colon mucosa (case 1).A: HE stain with diffuse atypical lymphocytic infiltration (original magnification ×50).B: Diffuse CD20 positive lymphocytes, immunohistochemistry stain (original magnification ×200).C: High Ki-67 index (original magnification ×100).D: Positive EBV infection,EBERin situhybridization stain cells stained positive for CD20,CD3 and Ki-67.e EBERin situhybridization stain was positive(original magnification ×200).
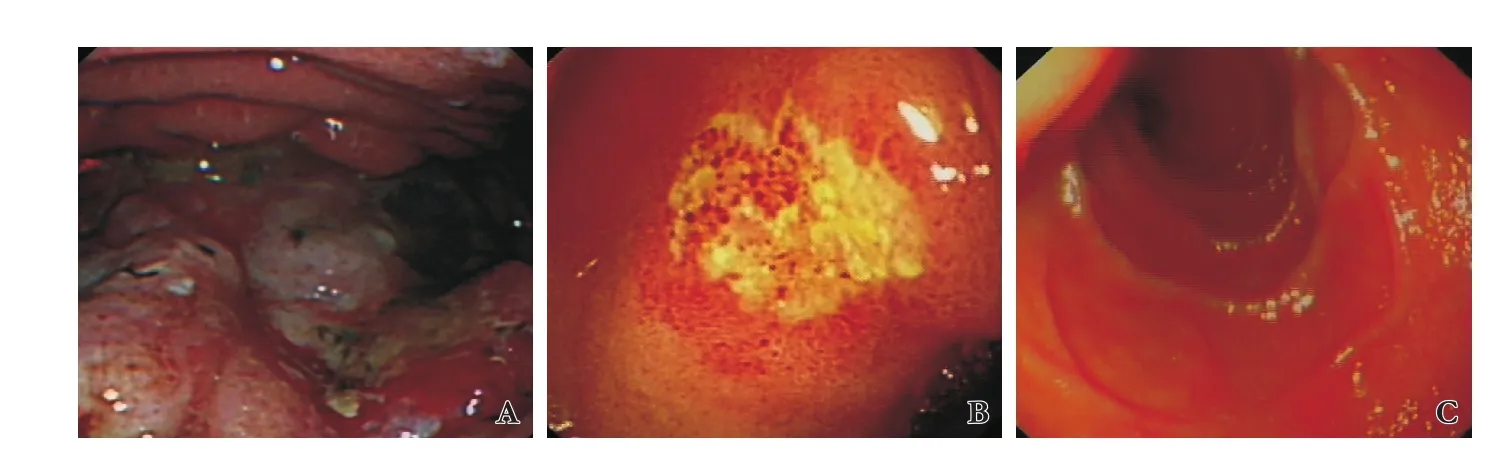
Fig. 2.Consecutive gastroscopy follow-ups (case 2).A: Gastric body large irregular ulcer at initial presentation.B: Gastric body ulcer aer 6-month CHOP therapy.C: Healed gastric ulcer at 36-month follow-up.
GI tract lesions represent a particular type of PTLDusually revealed by fever, abdominal pain (related to obstructive tumor mass or perforation) or upper or lower intestinal bleeding. Endoscopy together with capsule endoscopic examination are key to make the diagnosis.Treatment consists of reduction or withdrawal of immunosuppression and specific chemotherapy consisting mostly of anti-CD20 antibodies (rituximab) administration and/or more heavy chemotherapy such as CHOP,COMP, COP-E and MOP. Surgical intervention may be necessary in cases of severe GI-bleeding or GI tract obstruction or perforation.e final treatment has to be decided in function of the clinical and biochemical evolution of these immunosuppressed patients. Aggressive surgery for PTLD (even symptomatic) is controversial in literature,[10,11]especially since the introduction of very effective chemotherapy regimen combined with major reduction, or even withdrawal of immunosuppression.e presented medico-surgical approach allowed to render all four GI-PTLD patients disease free aer a follow-up ranging from 27.5 to 139 months.
Qin-Fen Xie, Ping Chen, Xin-Hua Chen, Ji-Min Liu,Jan Lerut and Shu-Sen Zheng
Department of Hepatobiliary and Pancreatic Surgery (Xie QF) and Department of Infectious Disease (Chen P), Shulan (Hangzhou) Hospital (Zhejiang University International Hospital), Hangzhou 310000, China; Key Laboratory of Combined Multi-organ Transplantation, Ministry of Public Health; Department of Hepatobiliary and Pancreatic Surgery, First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou 310003, China (Chen XH and Zheng SS); Department of Pathology and Molecular Medicine, Faculty of Health Science, McMaster University, Hamilton, Ontario, Canada (Liu JM); Starzl Unit of Abdominal Transplantation, Université catholique Louvain(UCL), Brussels, Belgium (Lerut J)
Corresponding Author: Shu-Sen Zheng
(Email: shusenzheng@zju.edu.cn)
Contributors:ZSS proposed the study. XQF performed the research and wrote the first dra. All authors contributed to the design and interpretation of the study and to further dras. ZSS is the guarantor.
Funding:None.
Ethical approval:is study was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University School of Medicine (2016-0484).
Competing interest:No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
1 Starzl TE, Nalesnik MA, Porter KA, Ho M, Iwatsuki S, Griffith BP, et al. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet 1984;1:583-587.
2 Penn I. Cancers complicating organ transplantation. N Engl J Med 1990;323:1767-1769.
3 Adami J, Gäbel H, Lindelöf B, Ekström K, Rydh B, Glimelius B, et al. Cancer risk following organ transplantation: a nationwide cohort study in Sweden. Br J Cancer 2003;89:1221-1227.
4 Rausch L, Koenecke C, Koch HF, Kaltenborn A, Emmanouilidis N, Pape L, et al. Matched-pair analysis: identification offactors with independent influence on the development of PTLD aer kidney or liver transplantation. Transplant Res 2016;5:6.
5 Petrara MR, Giunco S, Serraino D, Dolcetti R, De Rossi A.Post-transplant lymphoproliferative disorders: from epidemiology to pathogenesis-driven treatment. Cancer Lett 2015;369:37-44.
6 Lauro A, Arpinati M, Pinna AD. Managing the challenge of PTLD in liver and bowel transplant recipients. Br J Haematol 2015;169:157-172.
7 Morscio J, Tousseyn T. Recent insights in the pathogenesis of post-transplantation lymphoproliferative disorders. World J Transplant 2016;6:505-516.
8 Lai YC, Ni YH, Jou ST, Ho MC, Wu JF, Chen HL, et al. Post-transplantation lymphoproliferative disorders localizing to the gastrointestinal tract aer liver transplantation: report offive pediatric cases. Pediatr Transplant 2006;10:390-394.
9 Khedmat H, Taheri S. Lymphoproliferative disorders in pediatric liver allograrecipients: a review of 212 cases. Hematol Oncol Stem Celler 2012;5:84-90.
10 Cruz RJ Jr, Ramachandra S, Sasatomi E, DiMartini A, de Vera M, Fontes P, et al. Surgical management of gastrointestinal posttransplant lymphoproliferative disorders in liver transplant recipients. Transplantation 2012;94:417-423.
11 Doria C, Marino IR, Scott VL, Jaffe R, Minervini MI, Zajko A, et al. Posttransplant lymphoproliferative disorders presenting at sites of previous surgical intervention. Transplantation 2003;75:1066-1069.
(doi: 10.1016/S1499-3872(17)60063-8)
Published online September 7, 2017.
Kayvan Mohkam
(Email: kayvan.mohkam@chu-lyon.fr)
10.1016/S1499-3872(17)60058-4)
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- The “Colonial Wig” pancreaticojejunostomy:zero leaks with a novel technique for reconstruction after pancreaticoduodenectomy
- Risk factors and managements of hemorrhage associated with pancreatic fistula after pancreaticoduodenectomy
- Tailored pancreatic reconstruction after pancreaticoduodenectomy: a single-center experience of 892 cases
- Helicobacter pyloriand 17β-estradiol induce human intrahepatic biliary epithelial cell abnormal proliferation and oxidative DNA damage
- Prospective comparison of prophylactic antibiotic use between intravenous moxifloxacin and ceftriaxone for high-risk patients with post-ERCP cholangitis
- Comparative study of the effects of terlipressin versus splenectomy on liver regeneration after partial hepatectomy in rats