有机摩擦改进剂与金属铁的吸附性能研究
2017-09-22武志强钟锦声
刘 琼,赵 毅,武志强,钟锦声
(中国石化石油化工科学研究院,北京 10083)
有机摩擦改进剂与金属铁的吸附性能研究
刘 琼,赵 毅,武志强,钟锦声
(中国石化石油化工科学研究院,北京 10083)
采用量子化学的密度泛函理论考察了摩擦改进剂极性基团与金属铁的相互作用。探讨了多元醇酯、酰胺、羧酸、胺、醇、甲酯6种极性化合物的结构特点,确定了可能作用的活性原子,并考察了其与金属发生化学吸附的可能性。结果表明:丙酸、丙酸酰胺、丙酸甲酯、丙酸甘油酯、丙胺与铁均发生了化学作用;丙醇与铁发生了物理作用。计算了5种有机摩擦改进剂极性基团与Fe原子的化学吸附作用能,吸附强度由高到低的顺序为:丙酸甘油酯≈丙胺>丙酸酰胺>丙酸≈丙酸甲酯>丙醇。
量子力学 摩擦改进剂 化学吸附 物理吸附
发动机运行工况复杂,各摩擦副的润滑状态各不相同。在流体润滑状态下,润滑流体可有效隔开摩擦副,此时可通过降低润滑油黏度来改善润滑效果;边界润滑条件下,润滑油液体层不能连续存在,在基础油中加入摩擦改进剂是改善边界润滑的合理途径。边界润滑通常发生在高负荷、高速条件下,此时,两金属面间距离很短,微凸体发生间接接触;随着未来发动机设计条件越来越苛刻,边界润滑所占比重越来越大。为进一步改善发动机的润滑性能,摩擦改进剂的研究日益重要。
目前,关于摩擦改进剂的报道主要集中于摩擦性能测试、作用机理的试验研究上[1-2],理论方面研究报道较少。而深入研究添加剂结构本质对添加剂性能及作用机理具有重要的理论和实际意义。摩擦改进剂主要通过极性端基在金属表面形成物理或化学作用吸附在金属表面,分子间通过范德华力作用或静电作用形成油膜润滑。润滑油膜的持久性由极性端基与金属表面的吸附强度决定。润滑油在金属表面的吸附性能对其润滑效果具有重要影响。
研究人员对不同类型摩擦改进剂与金属表面的相互作用进行了考察。Kajdas等[3]研究了羧酸的摩擦化学反应;Tan等[4]研究了减摩剂醇、羧酸、甲酯与金属铝表面的相互作用。Tingle[5]通过脂肪酸边界润滑条件下的试验研究了脂肪酸的化学吸附性能。但对于摩擦改进剂与金属表面相互作用的考察尚不够深入。本研究采用量子力学方法进一步分析极性端基的吸附细节,探讨其结构与减摩性能的关系,希望为设计性能更好的摩擦改进剂提供有用的信息。
1 计算思路和方法
本研究采用Material studio 6.0软件包中的Visualizer模块构建初始构型,摩擦改进剂主要通过极性基团与金属表面形成吸附作用,因此为了简化计算,在可允许范围内以碳链中含3个碳原子的酸、甲酯、胺、酰胺等极性化合物为模型,模型结构见图1。采用基于密度泛函理论的量子力学从头计算模块Dmol3模块对摩擦改进剂模型化合物进行结构优化,对优化后的分子进行结构分析,判断其可能与金属Fe发生相互作用的活性原子;单个分子与金属铁表面的化学吸附作用实质上是分子中的杂原子向具有半空轨道——d轨道的铁提供孤对电子形成配位键,因此本研究进一步考察摩擦改进剂分子活性原子与铁发生化学作用的可能性,通过单个分子与铁原子的键合强度来考察油膜吸附的稳定程度,推测其减摩性能。吸附能Eads计算式为:
Eads=Emol/met-(Emol+Emet)
(1)
式中:Emol/met为添加剂与金属原子吸附体系的总能量,kJ/mol;Emol、Emet分别为添加剂和金属原子的总能量,kJ/mol。
在Dmol3计算中选用DND基组,GGA-PW91函数,收敛精度为:能量0.05 kJ/mol;受力1012N;位移5×10-13m。根据频率分析优化构型,无虚频则说明为体系能量最低构型。
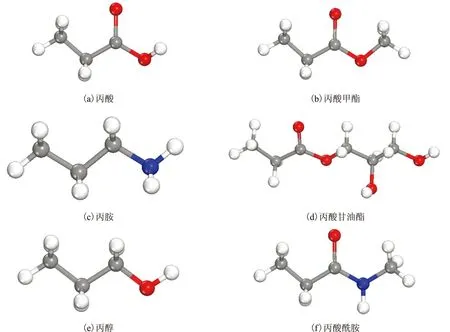
图1 不同极性基团摩擦改进剂的结构示意○—H; ●—C; ●—O; ●—N。图2~图5同
2 结果与讨论
2.1 基态分子结构讨论
分子前线轨道理论认为,分子中的电子按能量从低到高占据分子轨道,已填电子的能量最高轨道称为最高占据轨道(HOMO),能量最低的空轨道为最低空轨道(LUMO);分子发生化学反应取决于这些前线轨道。HOMO轨道集中位置即分子易发生给电子作用的活性位,LUMO轨道集中位置即分子易接受电子作用的活性位[6]。首先以优化结构的HOMO轨道来分析最可能发生供电作用的位置,图2为6种模型化合物的HOMO轨道图。从图2可以看出,丙酸的HOMO轨道主要集中在双键氧、单键氧和与羰基相连的碳上,说明这些原子最有可能与铁发生相互作用;丙胺的HOMO轨道主要集中在与氮和胺基相连的碳原子上;丙醇的HOMO轨道主要集中在羟基氧和与羟基相连的碳上;丙酸甲酯的HOMO轨道主要集中在双键氧、单键氧和与羰基相连的碳上;丙酸甘油酯的HOMO轨道主要集中在双键氧、单键氧和与羰基相连的碳上;丙酸酰胺的HOMO轨道主要集中在双键氧、氮和与羰基相连的碳上。
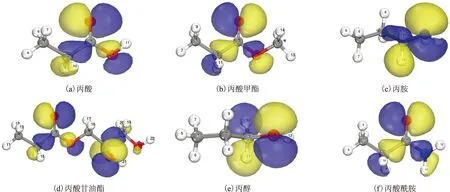
图2 不同极性基团摩擦改进剂的HOMO轨道
(2)
(3)
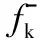
表1 不同极性基团摩擦改进剂分子中部分原子的指数
注:原子编号与图2中对应。
2.2摩擦改进剂与铁的相互作用考察
根据以上分析结果,进一步考察模型化合物分子中的活性原子与单个Fe原子的相互作用。分子发生反应时,一个分子的HOMO轨道和另一个分子的LUMO轨道必须对称性合适,互相起作用的HOMO轨道和LUMO轨道能级必须接近(约6 eV内)[7]。因此,通过模型化合物分子间轨道对称性来判断是否具有发生化学作用的可能性,通过成键轨道和是否有电子转移来判断其是否发生化学作用。以丙酸为例,首先分析丙酸与Fe发生作用的轨道。图3为丙酸与Fe作用的轨道示意。从图3可以看出,丙酸的HOMO轨道与Fe的空轨道能级相近(能级差6 eV以内)、对称性匹配,可发生化学作用。使Fe从羰基上方进攻丙酸,对此体系进行计算优化,对优化后的结构进行分析,可找到与之相应的成键轨道,如图3(a)所示;同时发现丙酸的LUMO轨道与Fe的占据轨道(HOMO)也发生相互作用,生成了相应的成键轨道,如图3(b)所示。分析原因,当Fe从羰基上方进攻时,一方面O给出孤对电子对与Fe的空轨道形成σ键;另一方面,Fe上的孤对d电子与丙酸的空轨道形成反馈π键,消除了金属原子上电荷的积累,使低价态的金属羰基化合物更加稳定。对结果进行进一步分析,当Fe从羰基上方进攻时,丙酸与Fe之间发生了电子转移,此时Fe上Mulliken电荷为0.185(见表2),C—O—Fe夹角约为79.710°,羰基氧与Fe间的距离约为0.196 9 nm,如图4所示。说明电子从Fe原子上转移到了羰基上,羧酸与Fe发生吸附后羧酸分子结构也发生了变化,C=O键、C—O键变长,表明羧酸与Fe发生了化学作用。
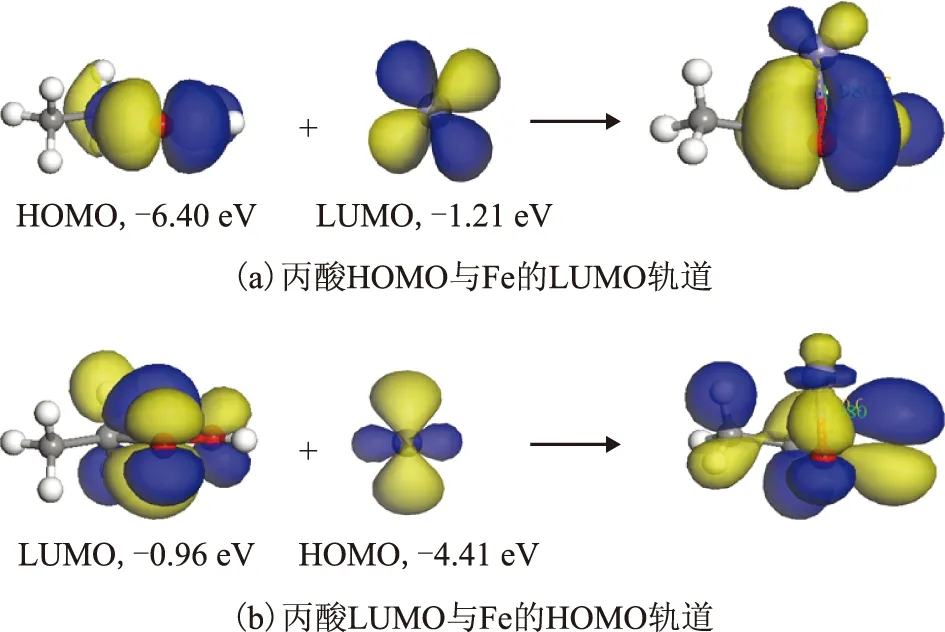
图3 丙酸与Fe作用轨道示意
用相同方法考察丙酸酰胺、丙酸甲酯、丙酸甘油酯、丙胺、丙醇与Fe的相互作用,发现丙酸酰胺、丙酸甲酯、丙酸甘油酯、丙胺与Fe作用时均发生了电荷转移,其与Fe的相互作用见图4,成键轨道见图5,表2为计算所得的化学吸附作用能(Eads)、优化后吸附末态作用原子与Fe间的距离(dads)、以及电荷转移情况。由图4、图5和表2可以看出,Fe与酰胺、甲酯、甘油酯的羰基均以与丙酸类似的方式发生配位作用,其中丙酸甲酯的C—O—Fe夹角约为83.157°,丙酸酰胺的C—O—Fe夹角约为81.503°,丙酸甘油酯的C—O—Fe夹角约为81.106°。丙醇由于只含电负性较弱的氧,不易给出电子,吸附过程中电子转移较少,且其吸附能只有-68.555 kJ/mol,认为丙醇只能与Fe发生弱的化学作用。
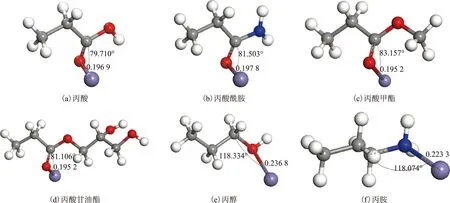
图4 摩擦改进剂活性原子与Fe作用的结构示意 键长单位:nm
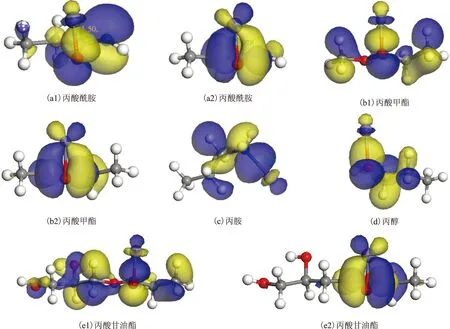
图5 丙酸酰胺、丙酸甲酯、丙胺、丙醇、丙酸甘油酯与Fe作用的成键轨道示意
从表2可以看出:吸附作用强度从高到低的顺序为:丙酸甘油酯≈丙胺>丙酸酰胺>丙酸≈丙酸甲酯>丙醇;对于烷基链相同、极性基团不同的摩擦改进剂,其主要差异在于极性基团;极性基团在Fe表面的吸附越强,形成的润滑油膜越稳定,减摩性能越好[8]。所以从吸附性能方面考虑,建议选用甘油酯、胺型等能与金属表面发生化学作用,且吸附稳定的摩擦改进剂,而醇类摩擦改进剂由于只能与金属表面发生弱的化学作用,吸附稳定性较差,在摩擦环境较苛刻的情况下,减摩效果可能不明显。
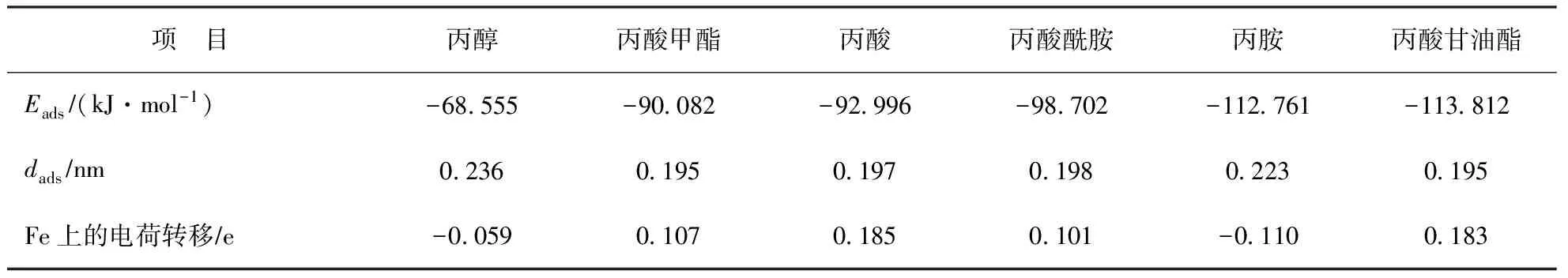
表2 不同极性基团摩擦改进剂与Fe的化学作用
3 结 论
(1)对不同极性基团摩擦改进剂模型化合物分子进行结构分析,认为丙酸、丙酸酰胺、丙酸甲酯、丙酸甘油酯与Fe发生相互作用的活性位为羰基,丙胺与Fe发生相互作用的活性原子为氮原子。
(2)采用量子力学模拟的方法研究了6种不同极性基团的摩擦改进剂与金属Fe的相互作用,结果表明:丙酸、丙酸酰胺、丙酸甲酯、丙酸甘油酯、丙胺与Fe均可发生化学吸附作用;丙醇与Fe只能发生弱的化学吸附作用;摩擦改进剂吸附强度从高到低的顺序为:丙酸甘油酯≈丙胺>丙酸酰胺>丙酸≈丙酸甲酯>丙醇。在筛选摩擦改进剂时,建议选用甘油酯、胺型等吸附稳定的摩擦改进剂,而醇类摩擦改进剂吸附稳定性较差,在摩擦环境较苛刻的情况下,减摩效果可能不明显。
[1] Bigelow W C,Brockway L O.Variation of contact angle and
structure with molecular length and surface density in adsorbed films of fatty acids[J].Journal of Colloid Science,1956,11(1):60-68
[2] Lundgren S M,Ruths M,Danerlöv K,et al.Effects of unsaturation on film structure and friction of fatty acids in a model base oil[J].Journal of Colloid and Interface Science,2008,326(2):530-536
[3] Kajdas C K.Importance of the triboemission process for tribochemical reaction[J].Tribology International,2005,38(3):337-353
[4] Tan Yuanqiang,Huang Weijiu,Wang Xueye.Molecular orbital indexes criteria for friction modifiers in boundary lubrication[J].Tribology International,2002,35(6):381-384
[5] Tingle E D.The importance of surface oxide films in the friction and lubrication of metals:Part I.The dry friction of surfaces freshly exposed to air[J].Transactions of the Faraday Society,1950,46:93-102
[6] Parr R G.Density-Functional Theory of Atoms and Molecules[M].New York:Oxford University Press,1989:99-104
[7] 周公度,段连运.结构化学基础 [M].3版.北京:北京大学出版社,2002:79-180
[8] Jahanmir S,Beltzer M.An adsorption model for friction in boundary lubrication[J].ASLE Transactions,1986,29(3):423-430
STUDYONADSORPTIONABILITYOFORGANICFRICTIONMODIFIERSONMETALIRONSURFACE
Liu Qiong, Zhao Yi, Wu Zhiqiang, Zhong Jinsheng
(SINOPECResearchInstituteofPetroleumProcessing,Beijing100083)
Quantum chemical density functional theory was used to study the interaction between the polar groups of friction modifiers(polyols,amide,carboxylic acid,amine,alcohol,and methyl ester)and metal iron.The structural characteristics of modifiers were studied to judge the active atoms of the modifiers and the possibility of chemisorption.The results show that the chemical action occurs between Fe and organic friction modifiers,such as propionic acid,propionate,propionate,propionate,propylamine.While physical action happens between propanol and Fe.The chemical adsorption energies were calculated between 5 kinds of polar groups and Fe.The intensity of adsorption is in the order:propionic acid glyceride ≈propylamine > propionic acid amide > propionic acid ≈methyl propanoate > propyl alcohol.
quantum mechanics; friction modifier; chemisorption; physical absorption
2017-02-14;修改稿收到日期:2017-03-16。
刘琼,硕士,工程师,从事润滑油产品的研发工作。
刘琼,E-mail:liuqiong.ripp@sinopec.com。
中国石油化工股份有限公司合同项目(110133)。
