矿物药白硇砂和紫硇砂的XRD、拉曼及近红外光谱特征研究△
2017-09-21明晶陈龙李娟陈科力黄必胜
明晶,陈龙,李娟,陈科力,黄必胜
(教育部中药资源和中药复方重点实验室 湖北中医药大学,湖北 武汉 430065)
·基础研究·
矿物药白硇砂和紫硇砂的XRD、拉曼及近红外光谱特征研究△
明晶,陈龙,李娟,陈科力,黄必胜*
(教育部中药资源和中药复方重点实验室 湖北中医药大学,湖北 武汉 430065)
目的:分析矿物药白硇砂和紫硇砂的X衍射(XRD)、拉曼(Raman)和近红外(NIR)光谱特征,明确其物相组成,为该两种药材及其易混淆品提供快速有效的鉴别方法。方法:采集白硇砂、紫硇砂共17批样品的XRD图谱,通过寻峰和物相检索确定其物相组成;在此基础上,分析样品的拉曼光谱特征,计算白硇砂和紫硇砂的平均光谱数值和相关系数,建立二者的拉曼光谱标准图谱。此外,还对二者的近红外光谱鉴别方法进行了初步探索。结果:XRD分析显示,白硇砂主要成分为氯化铵,尚含较多氯化钠;紫硇砂主要成分为氯化钠。同种药材之间,拉曼光谱相似度较高(相关系数均大于0.9)。此外,白硇砂近红外光谱特征峰明显且峰型较好,紫硇砂则无显著特征峰。结论:XRD分析可快速确定矿物药白硇砂和紫硇砂的物相组成;二者拉曼光谱具有良好的专属性,适于建立标准对照光谱特征图。白硇砂和紫硇砂的光谱特征的建立,为今后安全有效地应用该药材提供了有效依据。
白硇砂;紫硇砂;X衍射;拉曼光谱;NIR;物相组成;标准光谱
中药硇砂记载于《新修本草》和《本草纲目》等古代医药著作中[1],其又是一味特色民族药,在藏药及其成方制剂中较为常用,具有消积软坚、破瘀散结之功效,主要用于内积饱胀、经闭症、目翳胬肉、赘疣及痈肿疮毒等证[2]。据文献记载[3],硇砂分为紫硇砂与白硇砂,前者为卤化物类矿物紫色石盐晶体,主要成分为氯化钠;后者为氯化物类卤砂族矿物卤砂的晶体或工业合成品,主要成分为氯化铵。由于矿物类中药在外部形态上相似度较高,且多制成粉末销售,因此,市场上的混淆品和伪劣品较多,与动、植物类中药相比,鉴定矿物类中药难度更大。例如,白硇砂、硼砂、白矾等白色系矿物药,原药材的性状特征差异并不明显,药材粉末则更加难以鉴别。
X射线衍射(XRD)技术是一种针对固体粉末样品测试的现代分析方法[4-6],通过对所得XRD图谱进行解析,可快速而准确地得知样品的组成成分[7]。XRD技术用于矿物类中药的研究,可准确地确定矿物基原,为其他分析技术的应用奠定基础。激光拉曼光谱技术基于光的非弹性散射,通过激光与物质的相互作用产生拉曼位移来提供分子振动的相关信息,具有无损、快速、样品量少等优点[8-9],且所得拉曼图谱具有较强指纹性[10-11]。近红外光谱技术是近年来发展迅速的一种分析技术,在矿物类中药鉴别方面已得到成功的运用[12-14]。故本文尝试应用XRD、拉曼及近红外光谱技术3种现代分析技术对矿物药白硇砂和紫硇砂的来源、组成和鉴定方法进行深入研究,结合上述3种光谱技术的优势,使分析结果准确而可靠。
1 仪器与材料
1.1仪器
X’PertPro型X射线粉晶衍射仪(荷兰帕纳科公司),MIDJade6.0分析软件,Origin7.5数据分析和绘图软件;i-RamanPlus-785H型便携式拉曼光谱仪(美国必达泰克公司,激光波长785nm,薄型背照式CCD阵列检测器,配光纤探头及BWSpec4光谱采集软件);Matrix-F型近红外光谱仪(德国Bruker公司),配有光纤探头测样附件,铟镓砷检测器及OPUS5.0光谱分析软件。
1.2材料
从全国各大药材市场购入白硇砂、紫硇砂药材共17批,其中白硇砂7批(编号:S1~S7),紫硇砂10批(编号:S8~S17)。各批药材均由湖北中医药大学生药教研室陈科力教授鉴别为正品。氯化钠(分析纯,批号:20150410),氯化铵(分析纯,批号:20141027)。
2 方法与结果
2.1XRD图谱采集及分析
2.1.1XRD图谱采集 取适量上述2种药材样品及化学试剂氯化钠和氯化铵,粉碎后过200目筛,在玛瑙研钵中研匀,置于X衍射仪样品板的凹槽中,压平,测量。入射光源为CuKα辐射,Ni片滤波,X管工作电压为40kV,电流为40mA;光阑系统为DS=SS=1°;RS=0.3mm。连续扫描方式,扫描速度为8°·min-1,2θ分辨率为0.02°,扫描范围为3~65°,采用超能探测器。
2.1.2XRD图谱分析 测量所得XRD图谱在MIDJade6.0分析软件中寻峰处理,并与国际粉末衍射标准联合会(theJointCommitteeonPowerDiffractionStandards)编辑出版的“粉末衍射卡片PDF卡片”比对,检索其物相组成。同时,观察图谱发现同类药材图谱一致性较好,故选取白硇砂、紫硇砂代表性样品各一批绘图,如图1。
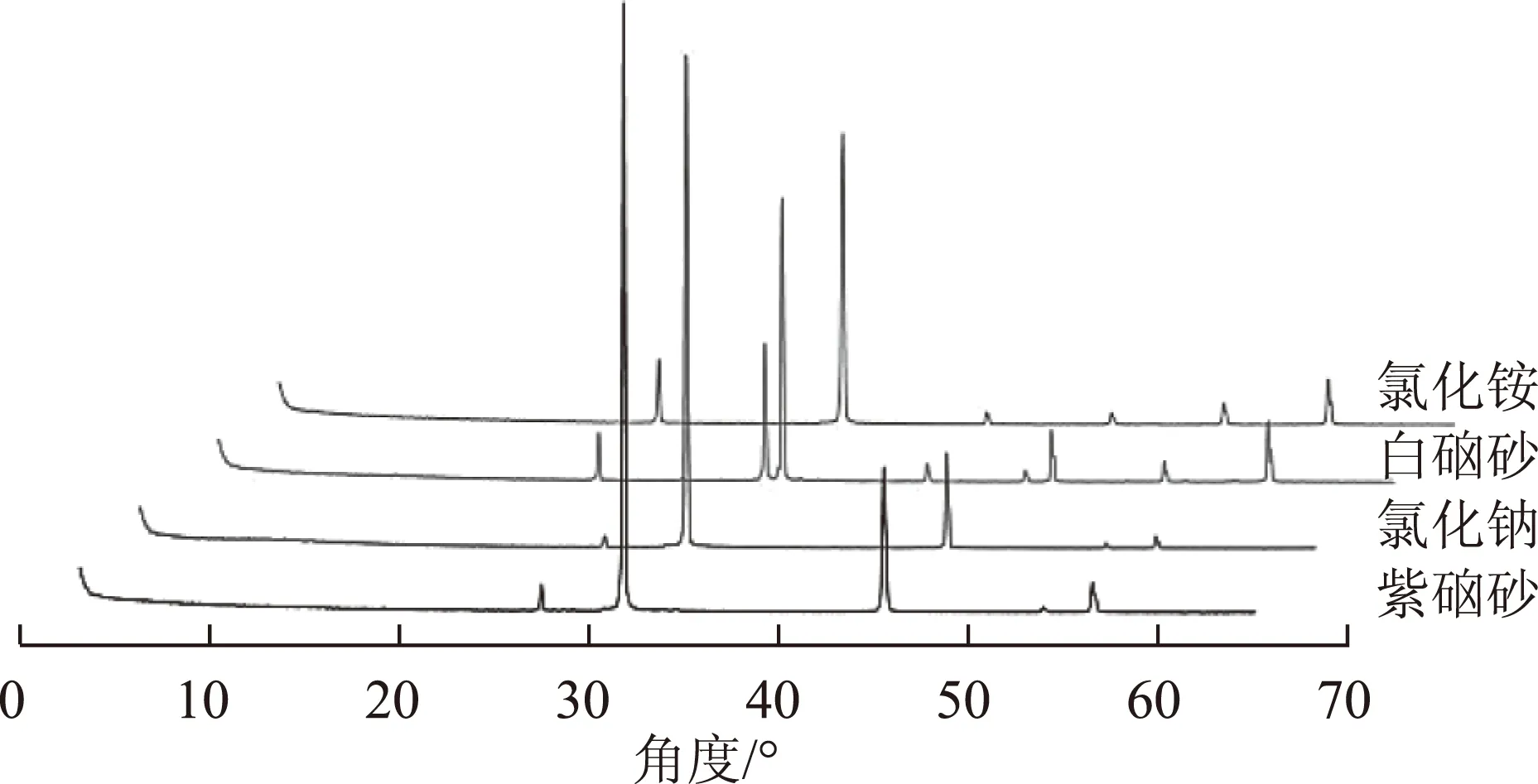
图1 白硇砂、紫硇砂、氯化钠及氯化铵XRD图谱
紫硇砂明显特征峰有5个,d(晶面间距,10-10m)=3.255,2.820,1.994,1.701,1.628,其中d=2.820为最强峰,d=1.994为次强峰,该组峰与氯化钠的特征峰大体一致,氯化钠的特征峰为:3.236、2.810、1.990、1.697、1.624。经过仔细对比,紫硇砂的特征峰与氯化钠的特征峰能够一一对应,但在峰位上具有一定的细微偏差,表现在紫硇砂的特征峰晶面间距均比氯化钠大0.004~0.02,导致这种现象的原因可能是晶体的结晶性差异。紫硇砂为天然形成的石盐,虽主要成分为氯化钠,但含有较多杂质成分分散在晶体中,且天然形成的石盐与工业合成的氯化钠晶体形成条件不同,自然结晶过程中的各种复杂因素(温度、湿度、压力等)导致紫硇砂的晶面间距整体增大。
白硇砂明显特征峰有9个,d=3.853、2.810、2.732、2.230、1.990、1.933、1.730、1.625、1.580,物相检索结果显示,其中d=3.853、2.732、2.230、1.933、1.730、1.580物相归属为氯化铵,d=2.810、1.990、1.625强度较弱,对应物相为氯化钠。据此可推断市售白硇砂不仅含有主要成分氯化铵,尚含部分氯化钠。
2.2拉曼光谱研究与标准特征图谱建立
2.2.1拉曼光谱采集方式和参数 紫硇砂为天然矿物,表面不规则,但经机械力敲击,能够产生平整而光滑、具有玻璃样光泽的断面,故将拉曼光纤探头轻抵新鲜断面采集光谱。而人工合成的白硇砂为细小颗粒压合而成的规则长方体,表面平整、均匀,可直接将探头轻抵样品洁净表面采集光谱。氯化铵和氯化钠试剂则取适量粉末置于样品杯中,压实,置于探头下测量。
拉曼仪器激发光源为785nm,光谱测量范围为2700~65cm-1,激光功率300mV,1.5m光纤探头,采用激光强度70%采集白硇砂样品氯化铵试剂光谱,扫描时间3000ms;采用激光强度100%采集紫硇砂样品及氯化钠试剂光谱,扫描时间60000ms,累积2次。每批样品采集3次求平均后进行Savitzky-Glay平滑,即得样品分析光谱。
2.2.2拉曼光谱分析7批白硇砂拉曼光谱如图2A,可见其一致性较好,具有5个特征峰,其中170cm-1和144cm-1左右为2个紧密相连的最强峰,93cm-1处有一强峰,还有2弱峰分别位于1708cm-1与1402cm-1处。通过查阅文献,93cm-1和144cm-1处两强峰由氯化铵晶体无序化诱发的振动模式产生[15],170cm-1处的峰对应氯化铵晶格振动模式[16],而1708cm-1处峰由氯化铵内部振动模式产生[17]。此外,有2批样品在1295、1128、1061cm-1处还存在3个极弱峰,与氯化铵试剂拉曼光谱对比发现,除了具有白硇砂的5个特征峰,氯化铵试剂亦有这3个弱峰。由此可推断,其余5批样品可能除了主要成分外,尚含较多其他成分,导致3个弱峰相对强度太弱而被噪声所掩盖,未被仪器检出。
由XRD物相分析,紫硇砂主要含氯化钠,为离子化合物,无共价键,故在拉曼图谱上无分子振动特征,其拉曼光谱特征性主要表现为晶格振动峰。10批紫硇砂样品拉曼光谱如图2B,可见其在(345±7)cm-1处有一最强峰且峰型较宽,在278、235、180cm-1左右分别出现3个峰,在412~380cm-1左右有1~2个弱峰,由于强度太弱,常被最强峰所掩盖。此外,部分样品图谱在597、459cm-1可见弱峰。
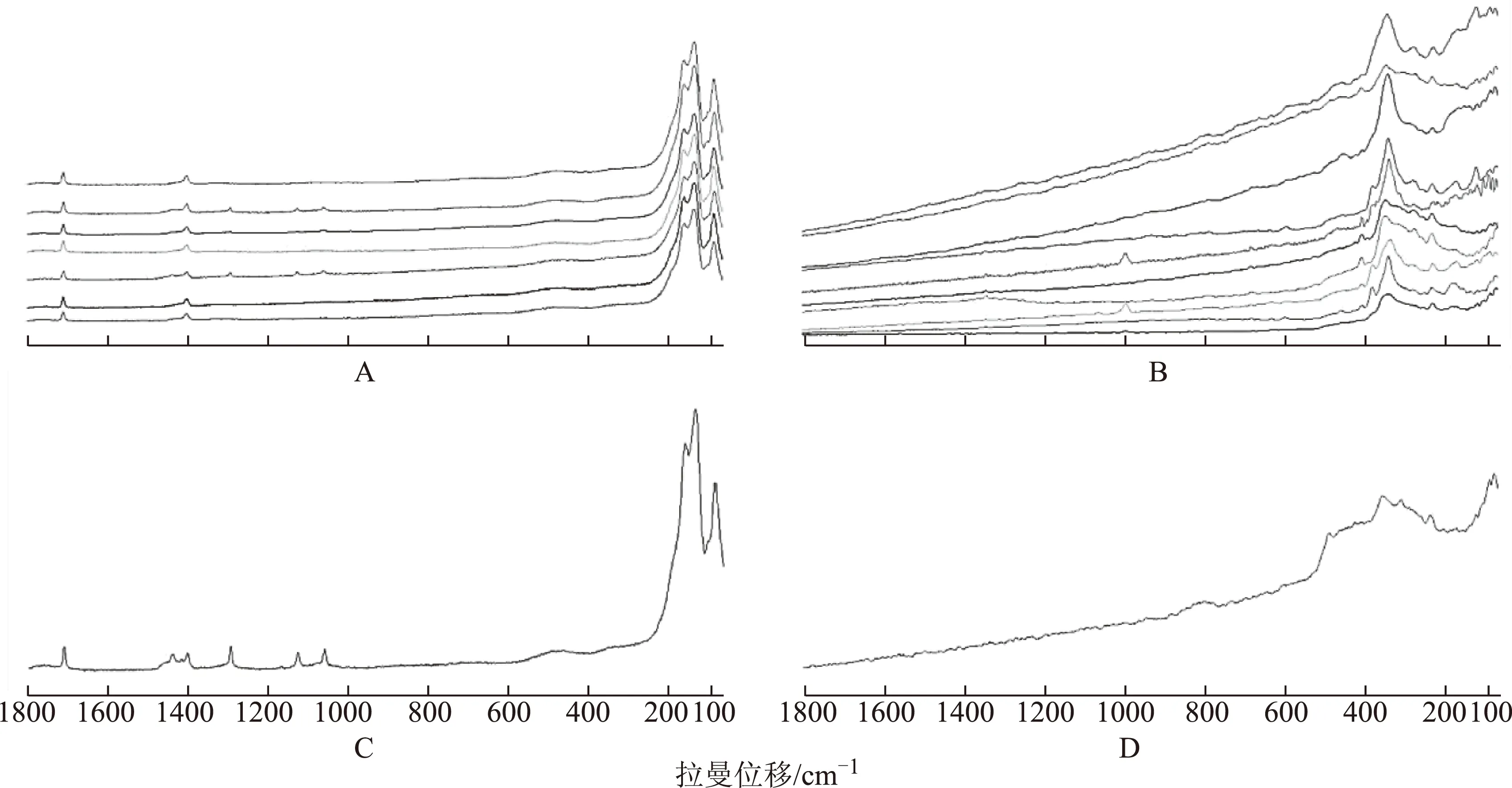
注:A.白硇砂;B.紫硇砂;C.氯化铵;D.氯化钠。图2 白硇砂与紫硇砂的拉曼光谱
2.2.3拉曼光谱标准特征图谱建立 通过白硇砂和紫硇砂样品拉曼光谱对比,同种药材光谱相似度高,而不同种药材具有较大差异,且二者特征峰主要表现在谱段1800~70cm-1,故选取这一谱段建立白硇砂和紫硇砂拉曼标准光谱。
采用叠加平均计算得到7批白硇砂样品与10批紫硇砂样品的拉曼平均光谱,分别计算白硇砂、紫硇砂样品拉曼光谱与所得平均光谱的相关系数。计算结果见表1。

表1 白硇砂与紫硇砂的拉曼光谱相关系数计算结果
相关系数计算结果显示,7批白硇砂样品拉曼光谱与白硇砂平均光谱相关系数均大于99%,与紫硇砂平均光谱相关系数均小于75%;10批紫硇砂样品拉曼光谱与紫硇砂平均光谱相关系数均大于90%,与白硇砂平均光谱相关系数均小于75%。该结果表明,白硇砂样品与紫硇砂样品拉曼光谱分别具有较高相似度,尤其白硇砂一致性更好,所得平均光谱特征性强,可作为紫硇砂、白硇砂药材的拉曼标准光谱,见图3。
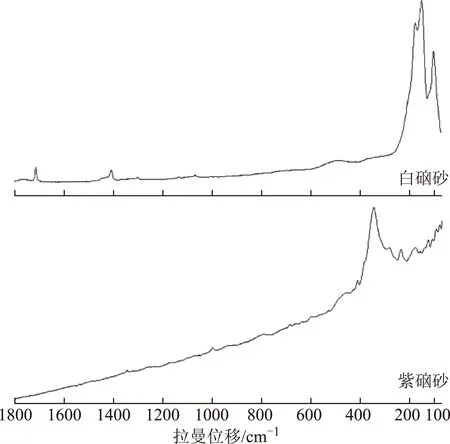
注:A.白硇砂;B.紫硇砂。图3 白硇砂和紫硇砂拉曼标准光谱
2.3近红外光谱采集与分析
2.3.1近红外光谱采集 取硇砂样品200目粉末置于样品杯中,压实,置于光纤探头下测量。采集参数为:扫描波数间隔8cm-1,扫描累积次数32次,扫描区间12000~4000cm-1。每批样品采集5张光谱,取平均光谱作为该样品的近红外光谱。
2.3.2近红外光谱分析 白硇砂与紫硇砂近红外光谱如图4,可见白硇砂近红外光谱出峰位置主要集中于谱段7000~4000cm-1,谱峰出现叠加现象,其中6250、5968、5850cm-13个峰叠加成为一宽峰,5140、4813、4560cm-13个峰叠加成为一强宽峰;此外,尚有一锐锋出现在4270cm-1处。白硇砂近红外光谱特征明显,适合用于近红外光谱分析。
由于近红外光谱主要反映含氢基团X-H(X=C、N、O)的振动[18-19],而紫硇砂的主要成分为NaCl,故其近红外光谱上除在5300~5000cm-1出现一水峰[14],其他区域未见明显特征吸收峰,谱线接近一条直线。故利用近红外光谱法分析鉴别紫硇砂存在一定局限性。

图4 白硇砂、紫硇砂、氯化铵及氯化钠近红外光谱
3 讨论
通过解析矿物药白硇砂与紫硇砂XRD图谱,可确定白硇砂与紫硇砂主要物相组成,明确二者矿物成分,为科学、合理用药提供了依据。所购白硇砂样品除含有文献记载的主要成分氯化铵外,尚含较多氯化钠。通过考察样品来源发现,市售白硇砂药材大部分为工业合成品,极少见天然矿物结晶,据此推断,白硇砂中含有的氯化钠成分可能与其工业合成方法有关,其药理药效是否与白硇砂天然矿物有明显差异有待进一步研究。
在XRD准确鉴定各个样品的基础上,通过分析样品的拉曼光谱特征,计算平均光谱以分别建立白硇砂和紫硇砂的拉曼光谱标准特征图谱,白硇砂和紫硇砂各个样品与其标准光谱的相似度均大于90%,表明该标准光谱可用于快速比对鉴别白硇砂和紫硇砂样品的真伪。其中,白硇砂样品与所建立的拉曼标准光谱相似度高,均大于99%;紫硇砂样品与拉曼标准光谱的一致性较白硇砂略低,在93.0%~99.7%,原因可能是白硇砂多为工业合成品;而紫硇砂多为天然结晶,在自然结晶过程中,由于温度、湿度、杂质等各种复杂因素的影响,不可避免导致紫硇砂结晶程度、所含杂质种类和含量存在一定差异。白硇砂和紫硇砂虽然颜色有明显差异,容易区别开,但与其同色系的其他矿物并不易区别。拉曼标准光谱为白硇砂和紫硇砂提供了一个可快速鉴别的光谱特征。
本文利用X射线衍射技术对白硇砂和紫硇砂进行成分分析确定其物相组成,在此基础上,进行了拉曼光谱特征分析,并建立了白硇砂和紫硇砂的拉曼标准光谱,还初步探索了白硇砂和紫硇砂的近红外光谱特征。这些工作运用现代光谱技术对矿物药白硇砂和紫硇砂进行了系统鉴别研究,为今后安全、有效地应用白硇砂和紫硇砂药材,提供了快速有效的鉴别方法。
[1] 江苏新医学院.中药大辞典:下册[M].上海:上海科学技术出版社,1988:2082.
[2] 国家中医药管理局.中华本草·藏药卷[M].上海:上海科学技术出版社,2002.
[3] 全国中草药汇编编写组.全国中草药汇编:下册[M].北京:人民卫生出版社,1978:537.
[4] 李树棠.晶体X射线衍射学基础[M].北京:冶金工业出版社,1990:131-148.
[5] 黄胜涛.固体X射线学(一)[M].北京:高等教育出版社,1985:197-224.
[6]HaroldPKlug,LeoryEAlexander.X-raydiffractionProceduresforpolycrystallineandamorphousmaterials(2ndEdition)[M].Wiley-Intersciencepublication,1974:505-553.
[7] 陈龙,袁明洋,雷咪,等.中药阳起石、阴起石的X衍射谱鉴别研究[J].中国中药杂志,2015,40(18):3560-3566.
[8] 李宁,庄正飞,郭周义,等.拉曼光谱技术及其在中医药鉴定中的应用[J].中国医学物理学杂志,2012,29(1):2173-3176.
[9] 陈龙,袁明洋,明晶,等.基于改进siPLS法建立5种含CaCO3中药的拉曼光谱定量模型[J].中国中药杂志,2015,40(18):3608-3615.
[10] 娄婷婷,王运庆,李金花.激光拉曼指纹图谱鉴别石油产品初探[J].光谱学与光谱分析,2012,32(1):132-136.
[11] 王文娜,陈地灵,朱梅芳.激光拉曼光谱法无损分析鉴别川贝母[J].光谱学与光谱分析,2013,33(8):2109-2111.
[12] 袁明洋.基于近红外光谱技术对几种矿物类中药的快速鉴别研究[D].武汉:湖北中医药大学,2014.
[13] 刘义梅,陈科力,袁明洋,等.近红外漫反射光谱法快速鉴别两组化石类中药材[J].世界科学技术—中医药现代化,2013,15(7):1538-1543.
[14] 袁明洋,黄必胜,余驰,等.8种含碳酸盐的矿物类中药近红外定性定量模型的建立[J].中国中药杂志,2014,39(2):267-272.
[15]YurtsevenH,KayisogluD.Criticalbehaviorofthedisorder-inducedRamanmodesclosetotheλ-phasetransitioninNH4Cl[J].SpectrochimicaActaPartAMolecular&BiomolecularSpectroscopy,2002,58(8):1823-1830.
[16]YurtsevenH,TümkayaMH.AnalysisoftheRamanfrequenciesofalatticemodeinNH4Clclosetothetricriticalandsecondorderphasetransitions[J].ChemicalPhysics,2006,323(2/3):574-578.
[17]YurtsevenH,TümkayaMH.RamanfrequencyshiftsofaninternalmodenearthetricriticalandsecondorderphasetransitionsinNH4Cl[J].SpectrochimicaActaPartAMolecular&BiomolecularSpectroscopy,2005,62(4/5):926-930.
[18] 翁诗甫.傅里叶变换红外光谱分析[M].北京:化学工业出版社,2010:359.
[19] 陆婉珍.现代近红外光谱分析技术[M].2版.北京:中国石化出版社,2000.
Study on XRD,Raman and NIR Spectral Characteristics of Sal Ammoniac and Halitum Violaceum
MING Jing,CHEN Long,LI Juan,CHEN Keli,HUANG Bisheng*
(Key laboratory of Traditional Chinese Medicine Resource and Compound Prescription,Ministry of Education & Hubei University of Chinese Medicine,Wuhan 430065,China)
Objective:To determine the original mineral resources and identification method of mineral traditional Chinese medicines Sal Ammoniac and Halitum Violaceum by analyzing their XRD Fourier patterns,Raman spectra and near infrared spectra(NIRS).Methods:XRD Fourier patterns collected from sample powders were used to determine phase composition.On this basis,Raman spectral characteristics were analyzed,average spectrum and correlation coefficient were computed,and thus standard Raman spectra of Sal Ammoniac and Halitum Violaceum were established.Besides,the identification method by NIRS was discussed preliminarily.Results:XRD analysis show that the main component of Sal Ammoniac was NH4Cl and Sal Ammoniac also contained much NaCl.The main component of Halitum Violaceum was NaCl.The Raman spectra of the same kind of medicinal materials exhibited a high similarity degree(the correlation coefficient was higher than 0.9).Besides,there were obvious characteristic peaks in the NIRS of Sal Ammoniac,while there was almost no obvious characteristic peak in the NIRS of Halitum Violaceum.Conclusion:XRD analysis is able to determine phase composition of Sal Ammoniac and Halitum Violaceum,and Raman spectra of both Sal Ammoniac and Halitum Violaceum have good specialization.It is appropriate to establish Raman standard spectrum,which could provide effective reference for the safe application of them.
Sal Ammoniac;Halitum Violaceum;XRD;Raman spectra;NIRS;phase composition;standard spectrum
重大新药创制国家科技重大专项(2014ZX09304307001);武汉市2012年高新技术产业发展行动计划生物技术与新医药专项(201260523193);湖北中医药大学校级教学研究项目(2015B15)
] 黄必胜,教授,研究方向:中药资源及其品质研究;E-mail:hbsh1963@163.com
10.13313/j.issn.1673-4890.2017.1.010
2016-05-04)
*[
