37例套细胞淋巴瘤患者临床病理特征及预后分析
2017-09-15宋锦添道光宁斌秀容
宋锦添, ,, , ,, ,道光,,宁斌,,,秀容,
(福建省肿瘤医院淋巴瘤内科,福建 福州 350014)
37例套细胞淋巴瘤患者临床病理特征及预后分析
宋锦添,杨瑜,施纯玫,陈强,吴晖,何鸿鸣,陈英,陈道光,林剑阳,陈宁斌,皱思平,王杰松,陈秀容,李梅芳
(福建省肿瘤医院淋巴瘤内科,福建 福州 350014)
目的探讨套细胞淋巴瘤(MCL)的临床特征、病理特点、预后因素,旨在提高对MCL的认识。方法回顾性分析了37例MCL患者的临床特征、病理特点、治疗过程及预后因素。结果37例MCL占同期我院收治的非霍奇金淋巴瘤的6.00%(37/617),中位年龄61(41~78)岁,发病高峰年龄段为50~70岁,男女比例为4.281。发病首发部位:多以浅表淋巴结肿大最为常见(75.68%),结外受累见于骨髓(45.94%)、脾脏(21.62%)、胃肠道(5.41%)等。初次就诊时处于Ⅱ期6例(16.22%)、Ⅲ期8例(21.62%)、Ⅳ期23例(62.16%),3组患者总生存时间分别为49.50、41.60、17.74个月,比较差异有统计学意义(P<0.05)。全组MIPI评分高危、中危、低危分别为16例(43.24%)、10例(27.03%)、11例(29.73%),3组患者总生存时间分别为45.43、42.4、21.32个月,比较差异有统计学意义(P<0.05)。全组中骨髓受累17例(45.95%),未受累20例(54.05%),未受累组预后较受累组好(总生存时间65.32个月vs22.12个月,P=0.010)。全组中,母细胞型14例(37.84%),经典型23例(62.16%),经典型组预后较母细胞型组好(总生存时间38个月vs18个月,P=0.028)。核分裂相<20/10 HPF 24例(64.86%),≥20/10 HPF 13例,核分裂相<20/10 HPF组预后较≥20/10 HPF组好(总生存时间50.34个月vs21.12个月,P=0.040)。结节型生长组23例(62.16%),弥漫型生长组14例(37.84%),2种生长方式对预后无明显影响(总生存时间28.33个月vs40.46个月,P=0.850)。全组病例共有36例行CD5检测,阳性35例(97.22%),阴性1例(2.78%);30例行CD10检测,阳性1例(3.33%),阴性29例(96.67%);22例行CD23检测,阳性5例(22.73%),阴性17例(77.37%);37例均行CyclinD1检测,阳性35例(94.59%),阴性2例(5.41%);20例行Ki-67检测,Ki-67≥50%组12例(60.00%),Ki-67<50%组8例(40.00%),Ki-67<50%组预后较Ki-67≥50.00%组好(总生存时间11.15个月vs31.62个月,P=0.001)。白细胞计数、淋巴细胞计数、乳酸脱氢酶水平、B症状等因素对生存预后无明显影响(P>0.05)。随访89(5~89)个月,随访率100.00%,生存超过2 a的共12例(32.43%),超过3 a的7例(18.92%)。结论MCL以全身浅表淋巴结肿大为常见首发症状,就诊时多已处于Ⅲ~Ⅳ期;CD5(+)、CyclinD(+)、CD10(-)、CD23(-)是常见的免疫学特征;分期晚、MIPI评分为高危、骨髓受累、母细胞变型、核分裂相≥20/10 HPF、Ki-67≥50%是预后不良因素;2 a生存率较低,预后较其他B细胞淋巴瘤相对差。
套细胞淋巴瘤;经典型;母细胞变型;预后
套细胞淋巴瘤(mantle cell lymphoma,MCL)是一种小B细胞非霍奇金淋巴瘤(non-Hodgkin lymphoma,NHL)。1992年WHO年首次将MCL作为一种独特的类型提出,2001将其划分为一种独立的NHL亚型,2008年在新分类和修订的欧美分类(REAL分类)中称之为套细胞淋巴瘤。与其他B细胞淋巴瘤对比,MCL具有惰性淋巴瘤的难治愈性和侵袭性淋巴瘤的侵袭性特征,故预后差。现将我院2008年10月至2014年10月收治的37例MCL患者的临床资料、病理资料、生存、治疗情况及预后因素进行分析,目的在于探讨MCL患者的临床、特征、病理特点、诊断、预后因素及治疗情况,以提高对本病的认识,为今后的研究及治疗提供参考。
1 资料与方法
1.1一般资料收集2008年10月至2014年10月福建省肿瘤医院收治的经病理组织学及免疫组化确诊的37例MCL患者的临床病理资料,收集资料包括:患者性别、年龄、发病时间、初诊时的病灶累及部位、B症状、病理组织学、免疫组化、分子生物学诊断、实验室检查结果、临床分期、套细胞淋巴瘤国际预后指数(mantle cell lymphoma international prognostic index,MIPI)、治疗情况等。
1.2实验室血液学指标包括白细胞计数、淋巴细胞计数、乳酸脱氢酶等。
1.3病理资料根据2008年WHO分类标准,对37例MCL的病理组织切片、免疫组化结果进行光镜下复习。观察内容包肿瘤细胞镜下形态、核仁大小及数目、染色质及胞浆,镜下病变模式、分型。其中病变模式分为套区增生型、结节型及弥漫型三种,其划分标准:套区增生型表现为残存生发中心被小至中等大小淋巴细胞样细胞增生形成的宽广外套层包绕,结节型在低倍镜下表现为肿瘤细胞呈模糊或清楚的结节状,且至少90%的结节已失去套区生长方式;弥漫型表现为均匀一致的、小至中等大小淋巴细胞样瘤细胞弥漫浸润[1]。当有2种病变模式并存时,以占50%以上的模式为准,并将MCL又分为经典型及母细胞变型。观察核分裂相,以10 HPF为单位,分为>20/10 HPF和≤20/10 HPF 2组。免疫组化收集指标包括:CD5、CD10、CD23、CyclinD1。
1.4分期标准收集病史、症状、体检、实验室检查、影像学、骨穿检查结果等资料,根据Ann Arbor-Cotswolds分期标准进行重新分期。
1.5MIPI评分根据MIPI评分将37例MCL进行评分,分为低危、中危、高危3组。MIPI评分标准见表2。

表1 MIPI评分标准
注:低危组:0~3分,中危组:4~5分,高危组:6~11分
1.6时间-事件参数的定义总生存期指自组织病理诊断明确之日起计算至任何原因死亡的时间或以末次随访日期为截尾日期。
1.7随访采用电话或信函方式进行随访,随访截止至2014年10月。

2 结果
2.1临床特征37例经病理确诊的MCL患者占我院同期收治的NHL 6%,其中男30例(81.08%)、女7例(18.92%),男女比例为4.291。发病中位年龄61(41~78)岁,发病高峰年龄段为50~70岁。以浅表淋巴结肿大为发病的首发部位者28例(75.68%),其中表现为颈部淋巴结肿大24例(64.86%),腹股沟淋巴结肿大17例(45.94%),腋窝淋巴结肿大16例(43.24%)。就诊时骨髓受累17例(45.95%),脾脏受累22例(59.46%),双肺受累1例(2.7%),胃肠道受累2例(5.41%),扁桃体肿大5例(13.51%)。按Ann-arbor分期,Ⅱ期6例(16.22%),Ⅲ期8例(21.62%),Ⅳ期23例(62.16%)。按MIPI评分,低危11例(29.73%),中危10例(27.03%),高危16例(43.24%)。伴有B症状11例(29.73%)。1例(2.70%)同时合并肺腺癌。
2.2实验室检查本组37例MCL患者初次就诊时白细胞计数升高有8例(11.63×109~159×109·L-1,占21.62%),中位值50.58×109·L-1,其中4位白血病样改变。淋巴细胞计数升高9例(6.19×109~98.32×109·L-1,占24.32%),中位值30.72×109·L-1。乳酸脱氢酶升高10例(245~704 u·L-1,占27.03%),中位值为329.14 u·L-1。
2.3病理组织学全组37例MCL患者活检病理组织镜下观察瘤细胞由小到中等大小且形态单一细胞组成。瘤细胞的细胞核不规则,染色质浓聚或较为分散,核仁多不明显,细胞质较少,细胞界限不甚清楚。细胞核形状虽然可不规则,但同一病例的细胞构成往往在形态上较为一致。核分裂相计数多少不等,多则可达30~40/10 HPF,多见于母细胞变型,少则3~10/10 HPF,多见于经典型。肿瘤生长方式可以呈套区状、结节状或弥漫型。全组病例中母细胞变型者14例(37.84%),经典型者23例(62.16%)。核分裂相>20/10 HPF共18例(48.65%),≤20/10 HPF共19例(51.35%)。套区+结节样生长23例(62.16%),弥漫型生长14例(37.84%)。
2.4免疫组化全组37例患者36例行CD5检测,其中CD5表达阳性35例(97.22%),表达阴性1例(2.78%);30例行CD10检测,其中CD10表达阳性1例(3.33%),表达阴性29例(96.67%);22例行CD23检测,其中CD23表达阳性5例(22.73%),表达阴性17例(77.27%);37例均行CyclinD1检测,阳性35例(94.59%),阴性2例(5.41%),但这2例FISH检测均存在t(11,14)(q13,q32)易位重排,且在病理形态学方面都符合MCL典型特征;20例行Ki-67水平检测,其中Ki-67≥50%组12例(60.00%),<50%组8例(40.00%),中位值为54.29%(20%~90%)。
2.5治疗及生存情况见表2。
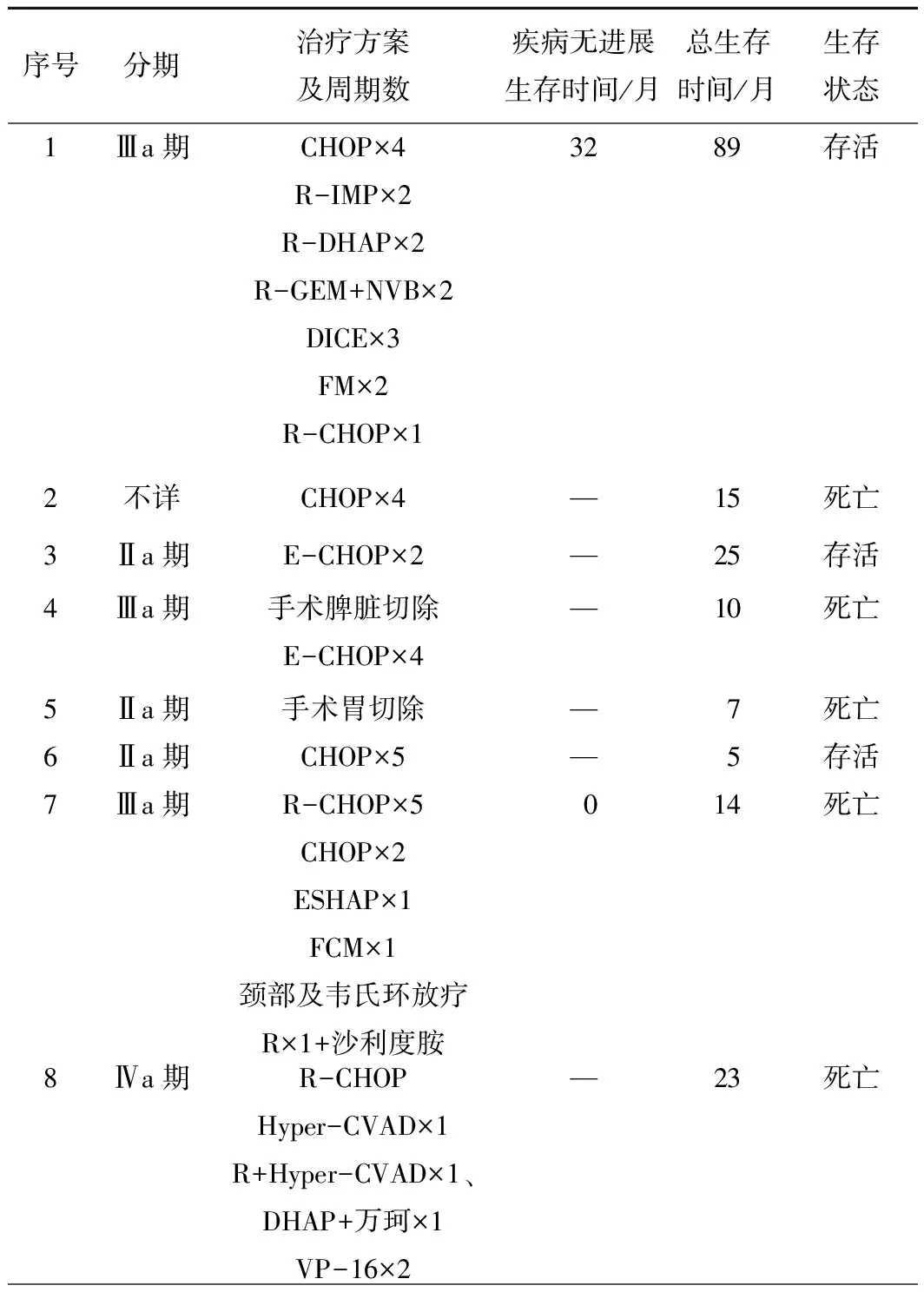
表2 37例患者的治疗及生存情况
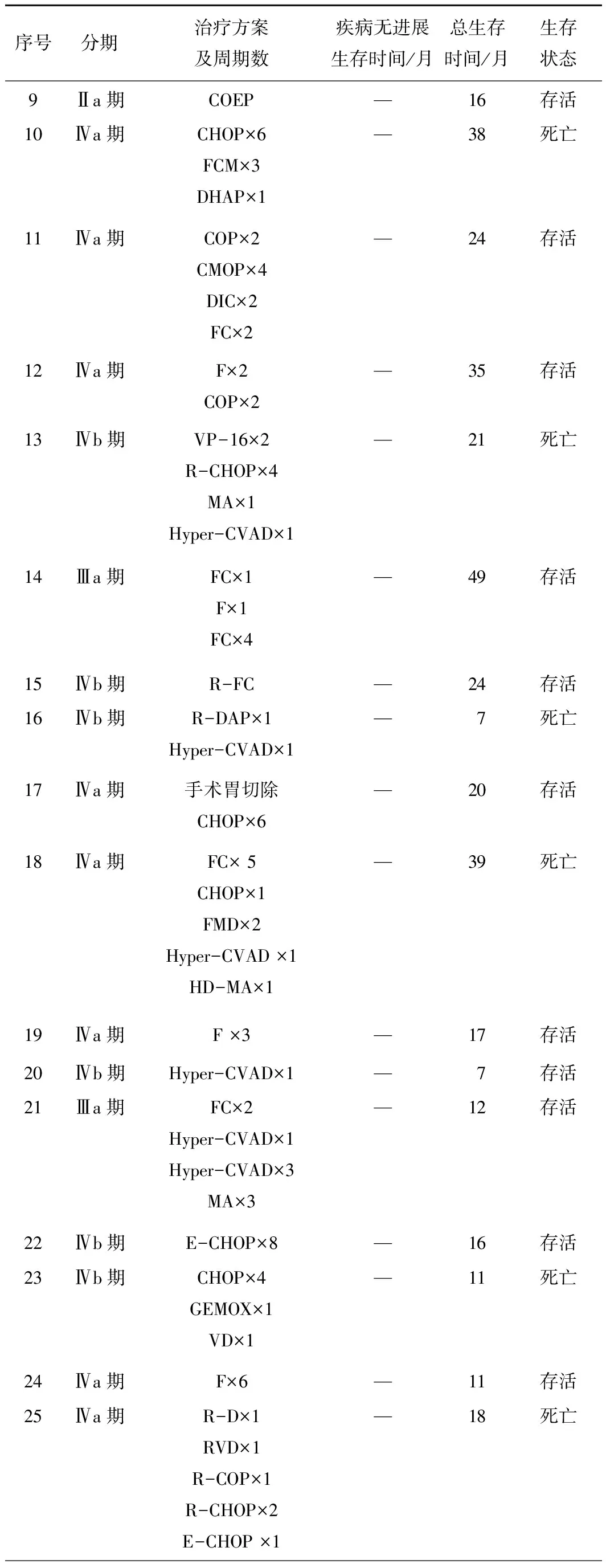
续表2

续表2
2.6生存分析
2.6.1 生存率分析 对37例患者性别、年龄、肿瘤分期、MIPI评分、骨髓受侵情况、病变模式、核分裂相、肿瘤生长方式、B症状、Ki-67水平、乳酸脱氢酶、白细胞计数、淋巴细胞计数等各因素累积生存率进行分析比较。见表4。

表3 各因素累积对37例MCL生存率的影响

续表3
2.6.2 COX回归分析 单因素分析:年龄、分期(Ann-Arbor分期)、MIPI评分、骨髓受累情况、瘤细胞病变型、核分裂相、Ki-67水平是套细胞淋巴瘤的预后影响因素(P<0.05)。COX回顾模型进行多因素分析却显示:以上因素均非预后影响因素(P>0.05)。

表4 套细胞淋巴瘤COX多因素分析
3 讨论
3.1诊断MCL是一种起源于淋巴结滤泡套区内小B细胞的侵袭性较强的NHL,诊断上有赖于依据病理组织学、免疫表型和细胞遗传学的检查。在组织病理形态学上,MCL常可分为经典型和母细胞样变型2种。经典型MCL镜下瘤细胞一般由形态单一的,小到中等大的淋巴样细胞构成。肿瘤细胞核形不规则,染色质浓聚或较为分散,核仁多不明显,细胞质较少,细胞界限不甚清楚。经典型MCL中一般见不到真正的转化大淋巴细胞。母细胞样变型在全部MCL中所占比例约6.3%[2]。在WHO分类中,母细胞样变型又分为经典型母细胞样变型和多形性母细胞样变型两中亚型。经典型母细胞样变型MCL瘤细胞形态较接近于淋巴细胞,细胞体积中等或偏大,染色质较细细致而分散,核仁不明显,核分裂相多见(至少10个/10 HPF,根据WHO分类诊断标准,需达到20~30个/10 HPF)。多形性母细胞样变型则以多形性大细胞增生为特点。肿瘤细胞的细胞核呈圆形或不规则,且至少部分细胞有明显核仁,细胞质也通常较为丰富[3]。因在组织处理不当或形态不典型时,MCL与其他小B细胞恶性淋巴瘤在形态上极易混淆,单凭HE形态学观察难以确诊[4],故此时免疫组织化学和遗传学的检测有助于正确的诊断和鉴别诊断。
MCL免疫表型特征为:SigM(+)、SIgD(+)、CD23(-)、CD43(+)、CD19(+)、CD20(+)、CD22(+)、CD79a(+)、CD74(+)、CD7(-)、CD3(-/+)、CD5(+)、CD10(-)、CD99(-)、CD34(-)、TdT(-),CyclinD1核蛋白(+),部分患者Bcl-6、c-myc、Ki-67在MCL细胞中阳性率高,其中CD5(+)、CD10(-)、CD23(-)是MCL特征免疫表型[5]。本全组37例患者36例行CD5检测,其中CD5表达阳性35例(97.22%),表达阴性1例(2.78%);30例行CD10检测,其中CD10表达阳性1例(3.33%),表达阴性29例(96.67%);22例行CD23检测,其中CD23表达阳性5例(22.73%),表达阴性17例(77.27%);37例均行CyclinD1检测,阳性35例(94.59%),阴性2例(5.41%),基本符合MCL免疫表型特征。
MCL大多数存在有t(11,14)(q13,q32)染色体易位[6],该易位使得位于14q32上编码CyclinD1的CCDN1基因与11q13上的IgH基因发生重排而靠近,导致CyclinD1蛋白持续的过度表达[7]。CyclinD1是一种重要的细胞周期调节因子,在控制细胞从G1期进入S期中起重要作用。故CyclinD1的过度表达会引起细胞周期性凋亡。免疫组化CyclinD1表达阳性是鉴别MCL与其他淋巴瘤的重要手段。本研究CyclinD1检测阴性2例行FISH检测均发现有t(11,14)(q13,q32)易位,在病理形态学方面都符合MCL典型特征。这对MCL都有CyclinD1的过度表达的观点提出了挑战。对CyclinD1表达阴性,但基因表达图谱研究却证实为MCL的病例,在蛋白水平上加以鉴别诊断难度较大。最近Fu等[8]又提出Y染色体性别决定区相关高速泳动族框因子11(SRY-related high mobility group-box genell,SOX11)可作为特异性标志物用于CyclinD1 阴性MCL患者的鉴别诊断。目前研究[9]表明SOX11除在MCL中具有强特异性核表达外,淋巴母细胞淋巴瘤/白血病、Burkitt淋巴瘤以及毛细胞白血病中可检测到SOX11,而在慢性淋巴细胞白血病/小淋巴细胞性淋巴瘤、滤泡型淋巴瘤、边缘区淋巴瘤以及弥漫大B细胞淋巴瘤及其他类型淋巴瘤中均为阴性,因此SOX11也被众多研究者推荐使用。
3.2临床表现MCL在中、老年人群高发,且有明显男性高发的特点。国外资料[10]显示,MCL好发于中、老年男性,中位年龄54~68岁,男女之比为1.6~6.81。1997年国际淋巴瘤组织在对NHL分类中报道提出MCL发病率占NHL的6%[11]。本组收集的37例MCL患者中,亦是以中、老年男性患者占多数,其中发病时中位年龄为61岁,男、女之比为4.281,占本院同期收治NHL的6.3%,这与国外报道的资料大致相符。
Matutes等[12]对836例MCL研究,结果显示80%~90%的患者确诊时疾病多已处于Ⅲ~Ⅳ期(Ann-Arbor分期),多数患者(>75%)表现有广泛性的淋巴结受累。大多数病例在发病时表现为缓慢无痛性进行性局部浅表淋巴结肿大。常见的结外受累部位为胃肠道、骨髓、Waldeyer环等,较少见的结外受累部位有皮肤,肺,乳腺等。约10%~15%的病例可能仅表现为结外病变而无淋巴结肿大,约30%~50%病例可有2个以上的结外部位累及。本组病例就诊时以浅表淋巴结肿大为首发表现共28例(75.68%),确诊时属Ⅲ~Ⅳ期共31例(83.78%)。22例发病时伴有脾肿大(59.46%),其中诊断为巨脾并伴有脾内瘤灶形成有7例(18.92%)。3例(8.12%)以胃肠道为原发病灶就诊,均经消化道内窥镜或手术病理证实。对于原发灶位于胃肠道的病例,多数表现为多发性的淋巴瘤性息肉病,常在肠道中有2个或2个以上病变部位,消化道内窥镜检查有高达88%的检出率,却仅有26%的患者有临床症状[13]。
Joshua等[14]曾报道MCL患者外周血累及呈白血病血相常见于至少1/4的病例,几乎全部病例流式细胞术可测出某种程度外周血累及,外周血受累是预后差的表现。本研究中白细胞计数升高有8例,其中有4例呈白血病样改变,中位生存时间为19个月,低于全组平均生存时间22个月并且4例均有骨髓侵犯。故推测白血病样改变的MCL预后较一般MCL预后差。
3.3预后因素迄今,许多学者都试图确定MCL具有预后意义的因素,但结果却不尽相同。国内有研究[15]曾对68例MCL患者的肿瘤细胞的病变模式、细胞类型、核分裂相进行过生存预后分析,其中母细胞变型较经典型预后差(P=0.042);≤15/10 HPF组较>15/10 HPF组预后好(P=0.003),套区增生型+结节型较弥漫型预后更好(P=0.006)。据Campo等[16]报道母细胞样变型患者起病较急,治疗情况较差,预后与经典MCL有明显不同。Bea等[17]曾分析了45例MCL患者(32例经典型和13例母细胞样变化)的染色体及DNA,结果发现母细胞变型患者的染色体异常及DNA扩增明显高于经典型患者,预后有显著差异,并证实母细胞变型、染色体和DNA的异常及临床预后之间有一定差异。而母细胞变型瘤细胞镜下则类似于淋巴母细胞,核分裂相常>20~30/10 HPF。对于母细胞变型病例,镜下核分裂相多>20/10 HPF,故两者在对患者预后方面结论较一致。本研究对以上3种因素进行了生存预后分析,得出结论如下:其中母细胞变型较经典型预后差(P=0.028)。核分裂相中以20/10 HPF为界限,≤20/10 HPF较>20/10 HPF预后好,2组生存曲线间差异有统计学意义(P=0.040)。病变模式中,套区增生型+结节型与弥漫型2组生存曲线对比无明显差异(P=0.085)。鉴于母细胞变型的较差预后,目前建议对MCL患者中,若病理证实为母细胞变型或核分裂相>20/10 HPF时,应明确地写在报告中。这对指导临床治疗及预后可能有意义。
Ki-67是一种非组蛋白性核蛋白,Ki-67的表达随细胞不同分裂阶段的改变而不同。因此可作为反映细胞分裂增殖状态的标志,用于评估肿瘤的生长增殖情况[18]。Ki-67与肿瘤的分化程度、浸润、转移情况及预后等关系密切。目前国外研究[19-20]显示Ki-67高表达与弥漫大B细胞淋巴瘤的不良预后相关。国内也有学者[21]曾对60例弥漫大B细胞淋巴瘤患者进行研究后发现,Ki-67在预后较好的生发中心来源亚型中低表达,而在预后较差的非生发中心来源亚型中高表达。而Ki-67的表达水平是否会对套细胞淋巴瘤的预后有指导意义,本文对Ki-67的表达水平与套细胞淋巴瘤的生存预后进行分析,结果提示Ki-67<50%组预后优于Ki-67≥50%组(P=0.001)。
Hoster等[22]在2008年依据MIPI评分分为低危、中危和高危三组,结论为:高危组预后最差,中危次之,低组较好(P<0.000 1)。本组资料按MIPI评分,低危组11例(29.73%),中危组10例(27.03%),高危组16例(43.24%)。3组患者的生存预后对比,低危组最好、中危组次之、高危组最差,生存曲线差异有统计学意义(P=0.045),故认为MIPI对MCL患者的预后估计和制定治疗方法有实际价值,可作为晚期MCL患者进行危险分层和个体治疗的指导工具。
据相关学者的研究[23-25]报道,MCL患者初诊时分期、骨髓是否受累及受累程度、白细胞计数升高、淋巴细胞计数升高、B症状、脾大、乳酸脱氢酶水平、贫血等因素对患者的预后均有影响,但结果并不一致。本文分别就肿瘤分期、骨髓侵犯、白细胞计数、淋巴细胞计数、乳酸脱氢酶水平、B症状、脾大等相关因素进行生存预后分析。结果显示:除分期晚、骨髓侵犯是预后不良因素外(P=0.004、0.010),其他因素对患者生存预后影响均不大(P>0.05)。
单因素分析提示分期、MIPI评分、骨髓受累情况、瘤细胞病变型、核分裂相、Ki-67水平、年龄均是套细胞淋巴瘤的预后影响因素(P<0.05)。而COX回顾模型进行多因素分析却显示:以上因素均与预后无明显相关(P>0.05)。
3.5治疗MCL兼有惰性淋巴瘤和侵袭性淋巴瘤的最差特征,常规的化疗不能治愈,但并没有惰性淋巴瘤的惰性病程,相反,其具侵袭性淋巴瘤较短的无进展生存期及总生存期。因MCL这种特殊的生物学行为,所以较其他小B淋巴细胞淋巴瘤预后差。淋巴瘤常用的CHOP方案不能有效控制该病,Joshua等[26]报道一线选用CHOP方案治疗MCL的总反应率约为75%,CR率仅为13%~50%。对于MCL目前仍无标准的一线治疗方案。最新美国NCCN指南提出的MCL一线治疗方案包括R-CHOP、R-HyperCVAD、R-EPOCH等,但是MCL对治疗反应较差,尽管患者最初对联合化疗有反应,但常在治疗1 a内复发。本研究因收集的病例数较少、治疗模式、方案差异大、治疗周期数在0~20周期不等并且缺乏治疗后疗效评估,故无法在统计学上进行性具体分析,仅能对部分病例治疗和生存情况做简要整理。全组37例病例有7例诊断后拒绝进一步治疗。其余30例病例中,分期为Ⅱ期共5例,治疗方面1例接受2周期E-CHOP方案化疗;1例接受5周期COEP方案化疗;1例接受5周期CHOP方案化疗;1例接受1周期CHOP方案及1周期Hyper-CVAD方案化疗;剩余1例仅单纯接受手术治疗。5例患者中除接受手术治疗的死于术后吻合口瘘外,其余4例随访至今均存活。存活4例患者平均年龄为68岁,MIPI评分属于高危组。故考虑对于Ⅱ期高龄患者,若无MIPI评分高危组等预后不良因素,可考虑较温和的CHOP样方案化疗。分期为Ⅲ~Ⅳ期共25例,接受化疗的方案各不相同。大体由Hyper-CVAD、CHOP、含氟达拉滨或以上方法联合利妥昔单抗治疗方案。25例病例中接受含有利妥昔单抗联合治疗的共10例,中位生存时间为23.5个月。15个例仅单纯接受化疗,中位生存时间18.6个月。故推测对于CD20(+)的Ⅳ期MCL患者,化疗联合利妥昔单抗较单纯化疗有更多获益。但因样本量较小且研究中化疗方案差异较大,周期数也不等以及其他混杂因素偏多(例如年龄、发病时间长短、男女性别比例等),故该推测尚且缺乏有效的说服性。在死亡的17例中,有4例因接受Hyper-CVAD/MA等强烈化疗方案,致Ⅳ度骨髓抑制伴严重肺部感染而死亡,4例患者中位年龄为69岁,故推测对高龄患者,强烈化疗方案不良反应明显,应慎重或减量使用。
对于化疗联合利妥昔单抗是否较单纯化疗有更大获益。Lenz等[27]比较122例初治MCL患者。随机应用CHOP与R-CHOP方案治疗,结果CHOP及R-CHOP方案的总有效率分别为75%和94%,CR率分别为7%和34%。另一项Ⅱ期临床研究[28]得出R-CHOP方案用于MCL的总有效率是96%,CR率为48%。可见利妥昔单抗联合CHOP方案对MCL患者的有效率较CHOP传统化疗方案有明显的提高。对于Ⅲ~Ⅳ期MCL患者,HyperCVAD方案是否较CHOP方案有更好的疗效,本研究也试图分析,因无确切的疗效评估,故无从得出结论。
MCL是一种少见的非霍奇金淋巴瘤亚型,多好发于中老年男性。大多数病例在疾病初期表现为浅表淋巴结肿大,且以颈部淋巴结肿大为常见。就诊时多已处于Ⅳ期(Ann-Arbor分期)。结外受累常见,其中常见的受累部位为骨髓、脾、外周血等。病理组织学上常可分为经典型及母细胞型,其中母细胞型又可分为经典母细胞型和多形型母细胞型。MCL的诊断有赖于病理组织学、免疫表型特征及细胞遗传学三者结合。免疫学表型方面CD5(+)、CyclinD1(+)、CD10(-)、CD23(-)是其特征型表现。细胞遗传学方面FISH检测到染色体t(11、14)(q13、q32)易位是诊断MCL的金标准。预后方面高龄、肿瘤分期晚、MIPI评分高危组、母细胞变型、核分裂相≥20/10 HPF、骨髓受累、Ki-67≥50%是预后不良因素。治疗方面尚无标准的一线化疗方案。常规的CHOP方案疗效差,强烈的Hyper-CVAD/MA化疗方案可提高疗效,血液学毒性却很明显。
[1] MAJLIS A, PUGH WC, RODRIGUEZ MA,et al.Mantle cell lymphoma: correlation of clinical outcome and biologic features with three histologic variants[J].J Clin Oncol,1997,15(4):1664-1671.
[2] ARGATOFF LH, CONNORS JM, KLASA RJ,et al.Mantle cell lymphoma: a clinicopathologic study of 80 cases[J].Blood,1997,89(6):2067-2078.
[3] 沈志祥,朱雄增.恶性淋巴瘤[M].2版.北京:人民卫生出版社,2011:595.
[4] 水若鸿,杨文涛,孙孟红,等.套细胞淋巴瘤石蜡包埋组织中细胞周期蛋白D1和t(11;14)易位检测的临床病理意义[J].中华病理学杂志,2003,32(4):340-341 .
[5] SINGH N, WRIGHT DH.The value of immunohistochemistry on paraffin wax embedded tissue sections in the differentiation of small lymphocytic and mantle cell lymphomas[J].J Clin Pathol, 1997 ,50(1):16-21.
[6] WILLIAMS ME, SWERDLOW SH, ROSENBERG CL, et al.Chromosome 11 translocation breakpoints at the PRAD1/cyclin D1 gene locus in centrocytic lymphoma[J].Leukemia,1993,7(2):241-245.
[7] MIGUET L, BÉCHADE G, FORNECKER L,et al.Proteomic analysis of malignant B-cell derived microparticles reveals CD148 as a potentially useful antigenic biomarker for mantle cell lymphoma diagnosis[J].J Proteome Res,2009,8(7):3346-3354.
[8] MOZOS A, ROYO C, HARTMANN E,et al.SOX11 expression is highly specific for mantle cell lymphoma and identifies the cyclin D1-negative subtype[J].Haematologica,2009,94(11):1555-1562.
[9] 曹鑫,范磊,方成,等.B淋巴增殖性疾病的SOX11、cyclin D1、cyclin D2和cyclin D3的表达研究[J].国际输血及血液学杂志,2011,34(1):4-9.
[10]VARDIMAN JW.The World Health Organization (WHO) classification of tumors of the hematopoietic and lymphoid tissues: an overview with emphasis on the myeloid neoplasms[J].Chem Biol Interact,2010,184(1/2):16-20.
[11]A clinical evaluation of the international lymphoma study group classification of non-Hodgkin’s lymphoma.The non-Hodgkin’s lymphoma classification project[J].Blood,1997,89(11):3909-3918.
[12]MATUTES E, PARRY-JONES N, BRITO-BABAPULLE V,et al.The leukemic presentation of mantle-cell lymphoma: disease features and prognostic factors in 58 patients[J].Leuk Lymphoma,2004,45(10):2007-2015.
[13]ROMAGUERA JE, MEDEIROS LJ, HAGEMEISTER FB,et al.Frequency of gastrointestinal involvement and its clinical significance in mantle cell lymphoma[J].Cancer,2003,97(3):586-591.
[14]BRODY J, ADVANI R.Treatment of mantle cell lymphoma: current approach and future directions[J].Crit Rev Oncol Hematol,2006,58(3):257-265.
[15]纪洪,李甘地,李俸媛,等.套细胞淋巴瘤的临床病理特征及预后因素分析[J].中华病理学杂志,2007,36(11):730-735.
[16]WEISENBURGER DD, VOSE JM, GREINER TC,et al.Mantle cell lymphoma.A clinicopathologic study of 68 cases from the Nebraska Lymphoma Study Group[J].Am J Hematol,2000,64(3):190-196.
[18]李迅,杨顺娥,赵兵.Bcl-6与Ki-67在非霍奇金淋巴瘤中的表达及临床意义[J].新疆医科大学学报,2009,32(8):1027-1031.
[20]YOON DH, CHOI DR, AHN HJ,et al.Ki-67 expression as a prognostic factor in diffuse large B-cell lymphoma patients treated with rituximab plus CHOP[J].Eur J Haematol,2010,85(2):149-157.
[21]宋恩霖,熊小亮,艾有生,等.Ki-67在GCB及non-GCB弥漫性大B细胞淋巴瘤中的表达及意义[J].江西医药,2011,46(2):111-114.
[22]HOSTER E, DREYLING M, KLAPPER W,et al.A new prognostic index (MIPI) for patients with advanced-stage mantle cell lymphoma[J].Blood,2008,111(2):558-565.
[23]熊竹娟,周祥,吴萍,等.21例套细胞淋巴瘤临床治疗及预后分析[J].肿瘤预防与治疗,2015,28(4):190-195.
[24]易树华,安刚,邹德慧,等.套细胞淋巴瘤27例临床特征及生存预后分析[J].中国实用内科杂志,2010,30(4):324-327.
[25]DETERMANN O, HOSTER E, OTT G,et al.Ki-67 predicts outcome in advanced-stage mantle cell lymphoma patients treated with anti-CD20 immunochemotherapy: results from randomized trials of the European MCL Network and the German Low Grade Lymphoma Study Group[J].Blood,2008,111(4):2385-2387.
[26]BRODY J, ADVANI R.Treatment of mantle cell lymphoma: current approach and future directions[J].Crit Rev Oncol Hematol,2006,58(3):257-265.
[27]LENZ G, DREYLING M, HOSTER E,et al.Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG)[J].J Clin Oncol,2005,23(9):1984-1992.
[28]HOWARD OM, GRIBBEN JG, NEUBERG DS,et al.Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival[J].J Clin Oncol,2002,20(5):1288-1294.
宋锦添(1985-),男,硕士,主要从事肿瘤内科临床工作。E-mail:songjintian0830@163.com
杨瑜(1962-)男,主任医师,主要从事淋巴瘤、晚期头颈部肿瘤、生殖细胞肿瘤的诊治工作。
10.3969/j.issn.1673-5412.2017.04.020
R735.9
B
1673-5412(2017)04-0334-08
2016-03-12)
