Fabricating Core-Shell WC@C/Pt Structures and its Enhanced Performance for Methanol Electrooxidation
2017-09-03ZhoyngChenLongDunYouqunChuJingfengShengWenfengLinChun
Zho-yng Chen,Long-f Dun,You-qun Chu,Jing-feng Sheng,Wen-feng Lin, Chun-n M
a.State Key Laboratory Breeding Base for Green Chemistry Synthesis Technology,College of Chemical Engineering,Zhejiang University of Technology,Hangzhou 310032,China
b.Department of Chemical Engineering,Loughborough University,Loughborough,Leicestershire,LE11 3TU,UK
Fabricating Core-Shell WC@C/Pt Structures and its Enhanced Performance for Methanol Electrooxidation
Zhao-yang Chena∗,Long-fa Duana,You-qun Chua∗,Jiang-feng Shenga,Wen-feng Linb, Chun-an Maa
a.State Key Laboratory Breeding Base for Green Chemistry Synthesis Technology,College of Chemical Engineering,Zhejiang University of Technology,Hangzhou 310032,China
b.Department of Chemical Engineering,Loughborough University,Loughborough,Leicestershire,LE11 3TU,UK
The spray-dried spheres within a W/Pt multi-separation can be used to prepare discrete core-shell WC@C/Pt catalysts through a typical carburization production mechanism at 800◦C.In contrast with previous studies of the WC/Pt synthesis,the reaction observed here proceeds through an indirect annealing mechanism at 600◦C wherein species diffuse, thereby resulting in core-shell structure,and Pt nanoparticles were successfully dispersed in size/shape and randomly scattered across the in situ produced C spheres.Through direct carburization or at higher initial hydrochloroplatinic acid concentrations,however,complete reaction with core-shell spheres was not observed.Indirect carburization reduces the strain felt by the bonds featuring the larger WC particles and allows the motion of carbon around WC and Pt nanoparticles to be reserved,in fluencing the electrocatalytic performance and stability toward methanol oxidation.
Self-disperse,Core-shell structure,Carbon-resist,Tungsten carbide,Methanol oxidation
I.INTRODUCTION
Pt-based catalysts demonstrate superior activity and stability for methanol electro oxidation in DMFC(Direct methanol fuel cells),and typical bimetallic catalysts include Pt-Ru,Pt-Au,and Pt-Pd alloys[1–4].Researches,in recent years,mostly focused on the costeffective and CO anti-poisoning improvement of Pt metals to accelerate the commercialization of DMFC[5–8].Pt nano-metalling,crystal plane control,and support effects are the main strategies to enhance the unit activity.The same synergistic effect strategy has also been extended to several other support effects systems.It is critical,therefore,to prepare high-efficiency precious metal-free supports(WC[9,10],MoC[11], and TiO2[12,13]),which is an effective way to reduce the Pt particle diameter and also increase the anti-poisoning ability.Tungsten carbide(WC)displays potential applications for Pt-like behavior[14],CO-tolerance[15],and highly electrical conductivity[16]as support part in anode-catalysts for methanol oxidation reaction(MOR)[17,18].Special crystal structures WC materials(porous WC[19–21],WC microspheres[22], nanochains[23],nanorods and nanoplatelets[24])have been prepared to enhance the electro-catalytic properties and stability for methanol oxidation.However, porous WC always exhibits coarse particle and smaller surface area than active carbon for its higher molecular mass.It has been researched to synthesize nano scale WC with a controllable size for achieving higher surface area,such as WC nanotubes with the diameter ranging from 30 nm to 70 nm[25,26].WC nanoparticles with an average size of 20 nm in diameter[27].Actually,the single WC particle is still difficult to meet the requirements for application in electrocatalysis.
The more active centers,therefore,must be effectively created by the nano WC-Pt contact of hybrid from construction of hierarchical pores rather than typical Pt doped WC particles.Furthermore,the coreshell hetero structures became an increasingly promising structure for the WC-Pt series catalysts,for it could effectively tailor the electronic structures which led to the dramatically enhanced catalytic ability and chemical stability[28].
Herein we reported a study in which the core-shell structure could be readily formed via a facile and costeffective method.More speci fically,Pt and WC structures have been successfully in situ fabricated and wellde fined on core-shell WC@C spheres.The as-prepared core-shell WC@C/Pt spheres manifest markedly enhanced electro catalytic performance and stability toward methanol oxidation.The reasons could be ascribed to the more active sites,the well mesoporousWC@C core-shell structures and synergistic effect between Pt catalyst and WC support.
II.EXPERIMENTS
A.Synthesis of the micro-sized spherical composites precursor(MSCP)
MSCP was prepared using spray drying(BCHI Spray Dryer B-290) treated an aqueous solution. Amixtureofammonium metatungstate (AMT,(NH4)2W4O13·xH2O)and chloroplatinic acid (H2PtCl6·6H2O,6.4 wt%Pt loaded vs.WC)were added to deionized water at 5 wt%. During spray drying process,the velocity of aqueous solution was 20 mL/min,the temperature of drying at the threshold and outlet was 200 and 100◦C,respectively.
B.Synthesis of the core-shell WC@C/Pt catalysts
The as-made MSCP underwent the process of calciation in air at 600◦C for 2 h,the obtained powder was then removed to a quartz tube,and was sent into tubal resistance furnace,which had been puri fied by N2for 0.5 h.Then this product underwent the gas-solid reaction under an atmosphere of CO/H2(125/250 cm3/min),the temperature was then increased to 800◦C and maintained for 4 h,the resulting product was denoted as indirect-carb WC@C/Pt. To be comparable,the precursor was directly carbonized without the heat treatment process under otherwise identical conditions,and the resulting product was denoted as direct-carb WC@C/Pt,H2PtCl6-free direct-carb WC was synthesized in the H2PtCl6-free directly carburization with other conditions same as direct-carb WC@C/Pt.
C.Characterization
The crystalline structures,composition,and morphology were investigated by X-ray diffraction analyzer(XRD,Rigakv D/max-III),scanning electron microscopy(SEM,Hitachi S-4800),high-resolution TEM using a Tecnai G2 F30 S-Twin microscope(FEI, Netherlands)at a voltage of 200 kV and a current of 103 mA coupled with energy dispersive X-ray spectrometer(EDX,Thermo NORAN VANSTAGE ESI). The thermogravimetric analysis(TGA)was performed on Pyris diamond thermoanalyzer.Particle-size distribution curve was performed on Malvern laser particle analyzer(MS2000).BET surface areas and pore size distributions were determined on a physical adsorption instrument(ASAP 2020,Micro-meritics Co.,USA).
D.Electrochemical measurements
Electrochemical measurements were carried out using a CHI600C electrochemical station,and a standard three-electrode setup with a saturated calomel electrode (SCE)and Pt foil as the reference and counter electrodes,respectively.For working electrode fabrication, indirect-carb and direct-carb WC@C/Pt(5 mg,respectively)were dispersed in a mixture aqueous solution of ethanol(1 mL)and Na fion solution(5 wt%,125µL)under ultrasonic treatment for 10 min.Afterwards,10µL of the catalyst ink(containing 0.044 mg of catalyst) were loaded onto a glassy carbon electrode(3 mm in diameter).Cyclic voltammetry(CV)was measured in N2saturated 0.5 mol/L H2SO4and 0.5 mol/L CH3OH solution at a scan rate of 50 mV/s.All the electrochemistry experiments were carried out at room temperature.The commercial Pt/C(20%Pt on Vulcan XC-72R,Johnson Matthey Corp.)and Pt black were also tested for comparison.
III.RESULTS AND DISCUSSION
SEM images of the MSCP are shown in FIG.1(a), hollow microsphere with thin wall(200−300 nm)were observed from the broken ones,the diameter of the microspheres was about 200 nm−5µm.It also can be found that the surface of hollow spherical was relatively smooth and there were an amount of spheres adhering to the surface both internal and external.
To investigate the factors contributing to the formation of the microsphere structure,TG-DTA patterns of the original AMT and MSCP are shown in inset of FIG.1(a).It can be speculated that the reaction of the original AMT(red line)in each stage along with the different temperature range is as follows:
(i)The mass of the AMT begins to decrease at the temperature from 150◦C to 230◦C,and weight loss comes to 3.85%of original weight.It is quite clear that the mass losses derive from free water and part of the crystalline water in original AMT,the equation is given by:
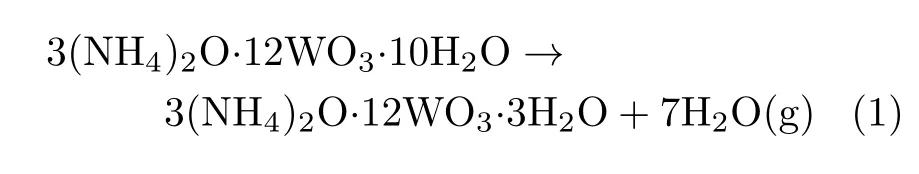
(ii)In the second step,decomposition between 290 and 340◦C leads to 5.01%weight loss,the resulting products are mainly ammonia,gaseous water and tungstate.The thermal decomposition reaction formula can be written as:
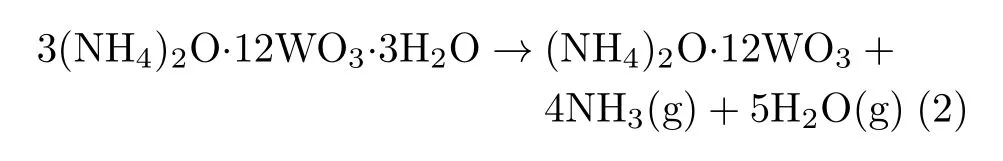
(iii)The weight of sample reduces slightly in the final decomposition around 390−410◦C,only results in 1.50%weight loss.This situation illustrates tungstatehas already decomposed into tungsten trioxide(WO3), as shown in Eq.(3):
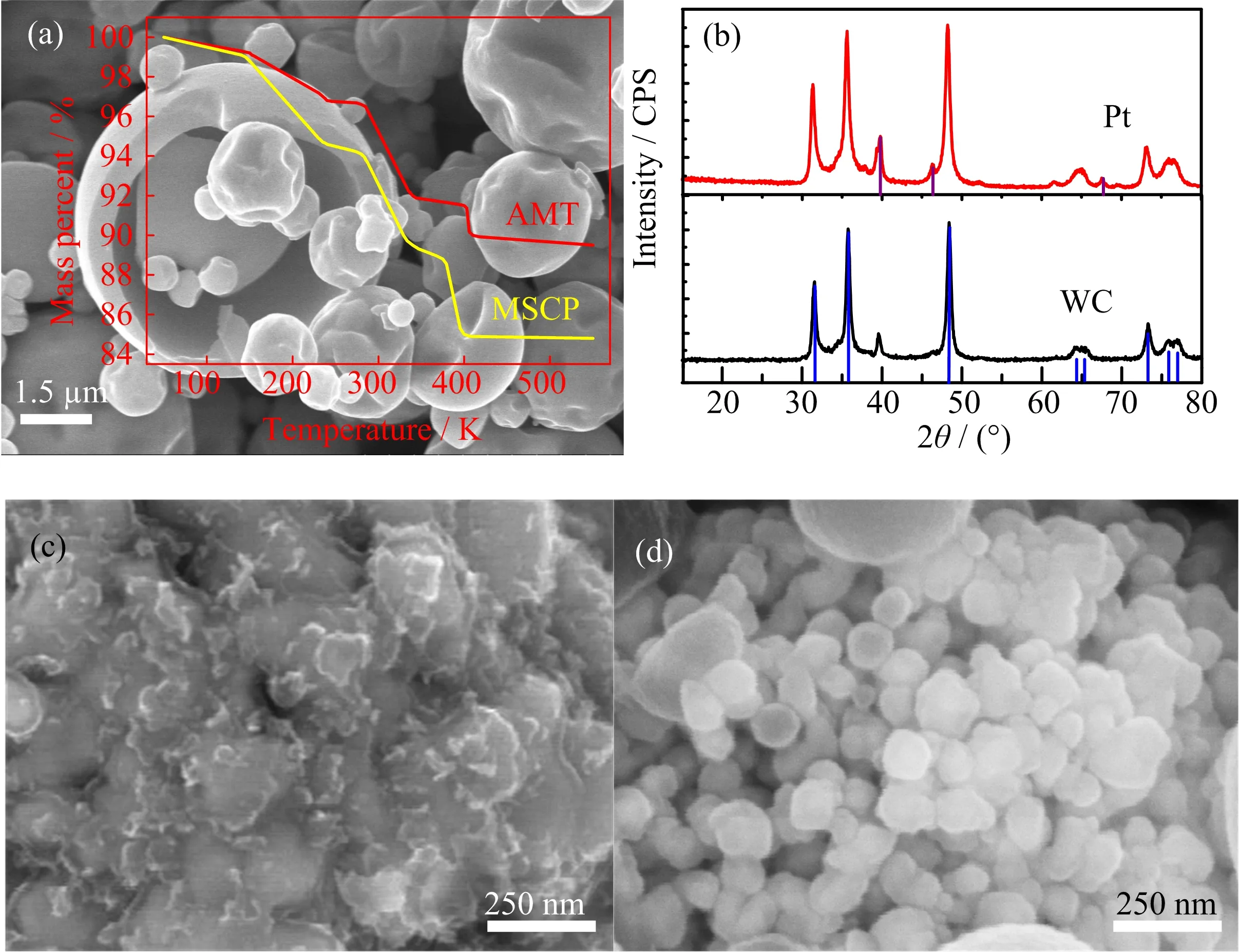
FIG.1(a)SEM images of the MSCP and TG curves(inset)of the original AMT and MSCP.(b)XRD patterns of direct-carb(black line)and indirect-carb(red line)WC@C/Pt composite.SEM images of(c)direct-carb WC@C/Pt and (d)indirect-carb WC@C/Pt composite.
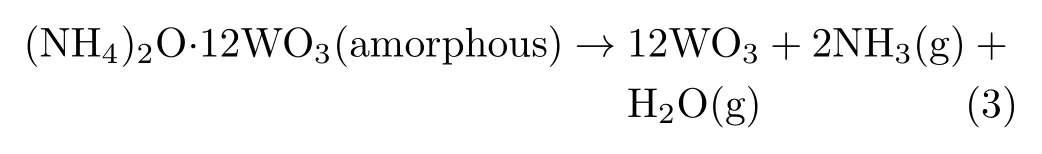
The AMT decomposition was completed and became stable when the temperature was over 450◦C, these results were consistent with the previous reports [29].As seen in FIG.1(a)(inset,yellow line),there are a lot of free water loss in the MSCP(weight loss take up 0.99%)from room temperature to 150◦C compared with original AMT.The crystal water of AMT and H2PtCl6are lost with the weight decreasing 4.55%in the temperature range of 150−255◦C.After that,AMT and H2PtCl6decomposed into tungstate and platinum tetrachloride(PtCl4),respectively,and the weight decreased about 5.50%in the range of 285−335◦C.Tungstate and PtCl4eventually decomposed into WO3,chlorine(Cl2)and platinum chloride (PtCl2)from 375◦C to 400◦C,respectively.In addition to the equation of AMT,the extra chemical reactions for MSCP could be formulated as follows:
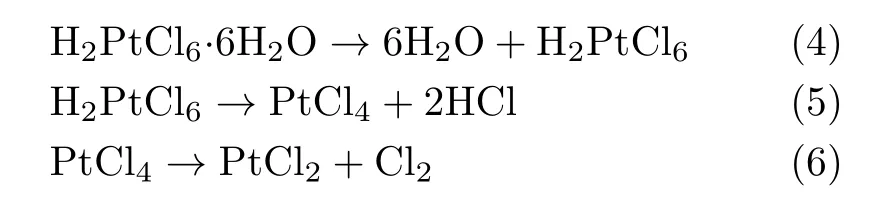
This information showed that Cl−concentration became lower during the heating process,and excessive reunion can be resisted. Thus,the heat treatment at 600◦C was employed to promote the formation of mesoporous structure and particles[30].The sample was prepared after annealing and carbonization process,and signed as indirect carb WC@C/Pt.By contrast,we also obtained the sample through direct carburation of the MSCP without 600◦C anneal process and signed as direct-carb WC@C/Pt. To analyze the phase structure,XRD patterns determinations were applied to characterize the obtained samples after the MSCP carburization.FIG.1(b)shows XRD pattern of the as-synthesized WC@C/Pt,where main peaks at 2θ=31.52◦,35.70◦,and 48.25◦correspond to diffractions from(001),(100),and(101) planes of hexagonal WC(JCPDS No.51-0939).Moreover,XRD pattern displays diffraction peaks at 39.79◦, 46.28◦,and 67.53◦correspond to the crystal planes of Pt(111),(200),and(220),respectively.The XRD data con firmed the formation of Pt nanoparticles with face-centered-cubic(fcc),which was consistent with the JCPDS No.87-0646.Nevertheless,compared with direct-carb WC@C/Pt,the indirect-carb WC@C/Pt exhibited stronger Pt diffraction peaks,especially(111) direction,which was due to the indirect carburization process in favor of the Pt growth prefering(111)direction.For Pt crystal,the surface energies associated with the low-index crystallographic planes were in the order of γ(111)<γ(100)<γ(110)[31].Carbon component,which was produced in the indirect carburization process,provided better macroscopic melting condition as the support of the Pt nano particles.During the high temperature treatment,the(111)plane of Pt in the indirect carburization sample was in dominant growth. The Pt particles in the direct carburization sample were covered by the WC and the lower surface energy plane was not in dominant growth.
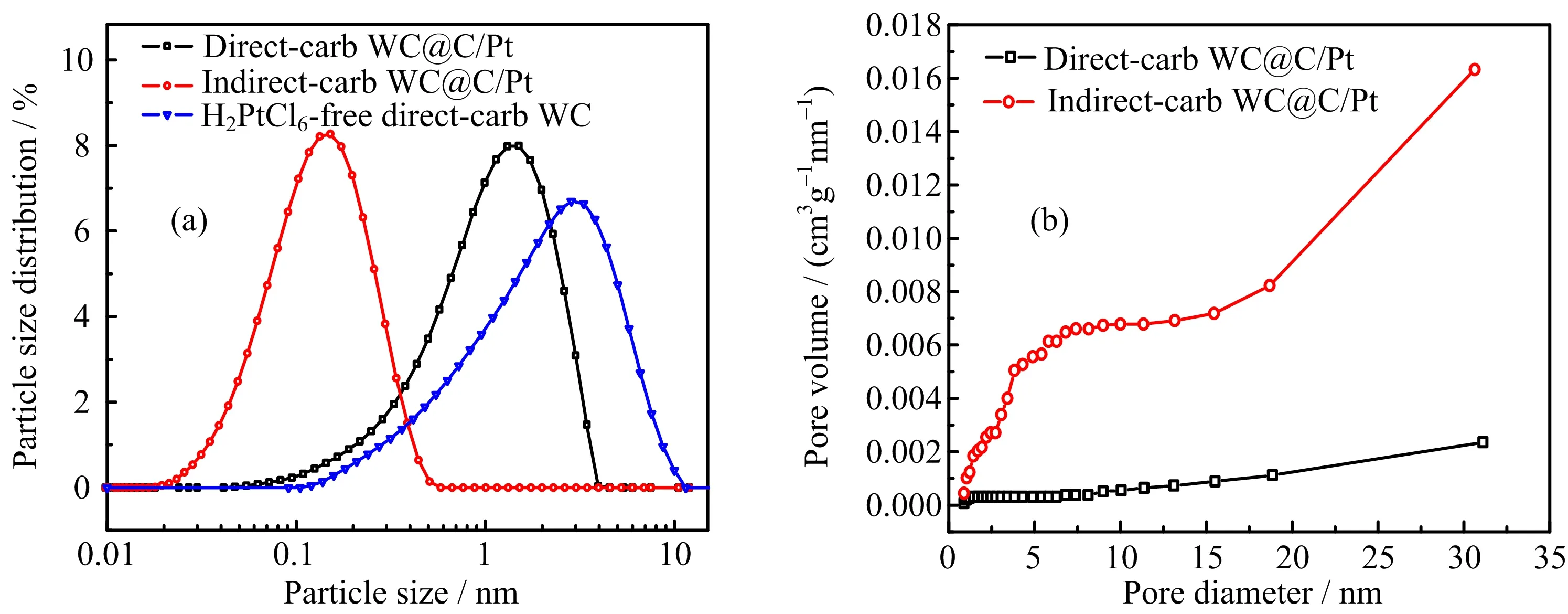
FIG.2(a)The particle size distributions of H2PtCl6-free direct-carb WC and direct/indirect-carb WC@C/Pt.(b)Pore size distributions for direct/indirect-WC@C/Pt sample.
The microstructures and morphologies for WC@C/Pt weredeterminedbySEM.From FIG.1(c),the WC@C/Ptobtainedbydirectcarbonizationwas severely agglomerated with coarse particles,on the contrary,the indirect-carb WC@C/Pt(FIG.1(d))displayed a remarkably spherical structure with diameters ranging between 50 and 120 nm.
The particle size distributions of direct-carb WC and direct/indirect-carb WC@C/Pt are given in FIG.2(a). Compared to H2PtCl6-free direct-carb WC sample,the particle size of direct-carb WC@C/Pt decreased slightly due to that the carburization with participation of Cl−led the particle to agglomerate,which finally promoted WC particles size to increase.However,particle size distributions of indirect-carb WC@C/Pt exhibited much smaller than direct-carb WC@C/Pt,resulted from the indirect-carburization processes which made the larger WC particles break and the secondary particles reduce greatly in size.The results indicated that the indirectcarburization processes promoted the ball to uniformly disperse and particles size to decrease during high temperature and carburization processes with Cl−participating.This phenomenon was much different from typical double heating treatments which inevitably became more highly branched and eventually entangled into large WC and Pt agglomerates.The discrepancies appeared to be related to the effect of chloride(Cl−)ions from H2PtCl6or PtCl2.In indirect carbonizing process, the crystal water,NH3,HCl,Cl2were removed and the mesoporous skeleton structure of MCSP was formed, it was bene ficial to Cl−ions(from PtCl2)removal and impeded the secondary reunion of WC and Pt nanoparticles for WC@C/Pt.We also added H2PtCl6in higher concentration(10 wt%and 15 wt%Pt loaded on WC), the MCSP particles were much larger than the original one,which was caused by the Cl−concentration and solution viscosity increase.And the spray-dried particles would not be well preserved.
Pore size distributions of different samples are given in FIG.2(b). The results indicate that indirectcarb WC@C/Pt exhibits a higher speci fic surface(11.687 m2/g)which is17.3 timesofdirect-carb WC@C/Pt(0.675 m2/g).Pore size distribution shows the indirect-carb WC@C/Pt has better mesoporous structures.It is clear that the higher surface area was resulted mostly from the mesopores even when there were micropores present.These mesopores formed partially because the carbons were in situ produced from the CO decomposition during the carbonization.
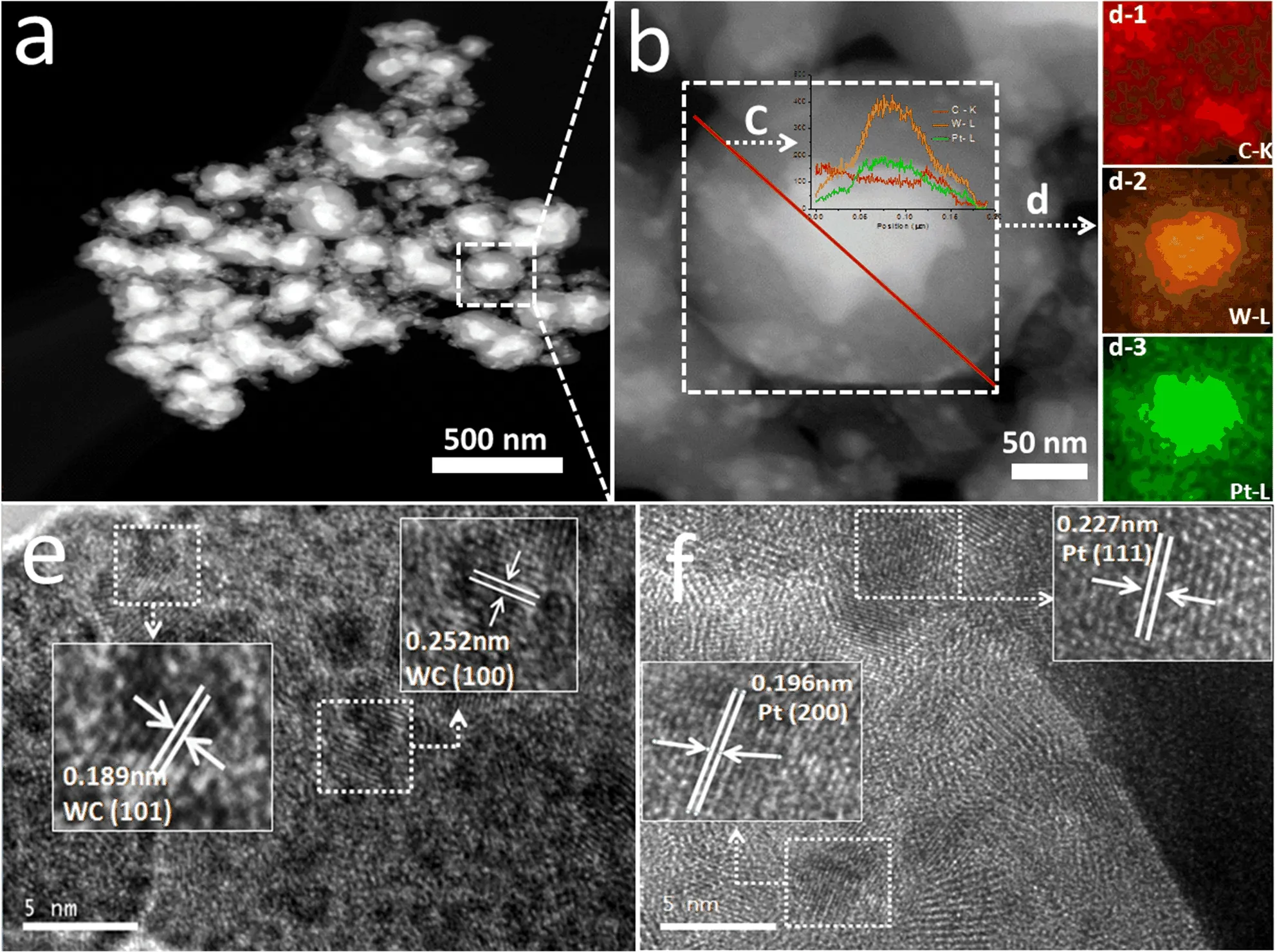
FIG.3(a,b)STEM images,(c)EDX line-scan analysis and (d1,d2,d3)elemental mapping,(e,f)HRTEM images of the indirect-carb WC@C/Pt composite.
FIG.3(a)−(f)shows the typical STEM and highresolution TEM(HRTEM)images of indirect-carb WC@C/Pt as well as their elemental distribution. Clearly,the material displays spheres with core-shell structure from FIG.3(a).The core-shell structure was further con firmed by EDX line-scan analysis along the selected line across the particle(FIG.3(b),red line).As shown in FIG.3(c),it showed distinctly the WC-core and C-shell structure for WC@C/Pt.
Moreover,in order to demonstrate the Pt nanoparticles can be well-dispersed on the WC support,elemental mapping had been performed and it proved C,W andPt were uniformly distributed and overlapped in the spheres(FIG.3(d1,d2,d3)).As shown in HRTEM image(FIG.3(e,f)),the crystalline lattices of WC(101) and(100),Pt(111),and(200)facets can be clearly observed,which is consistent with the results of XRD analysis.It shows that WC with mesoporous carbon shells on which WC and Pt particles uniformly dispersed have been prepared.Pt and WC thus successfully were separated without more impregnation or high-temperature hydrogen-reduction processes.
FIG.4(a)EDX spectrum and(b)TG-DTA curves of indirect-carb WC@C/Pt.
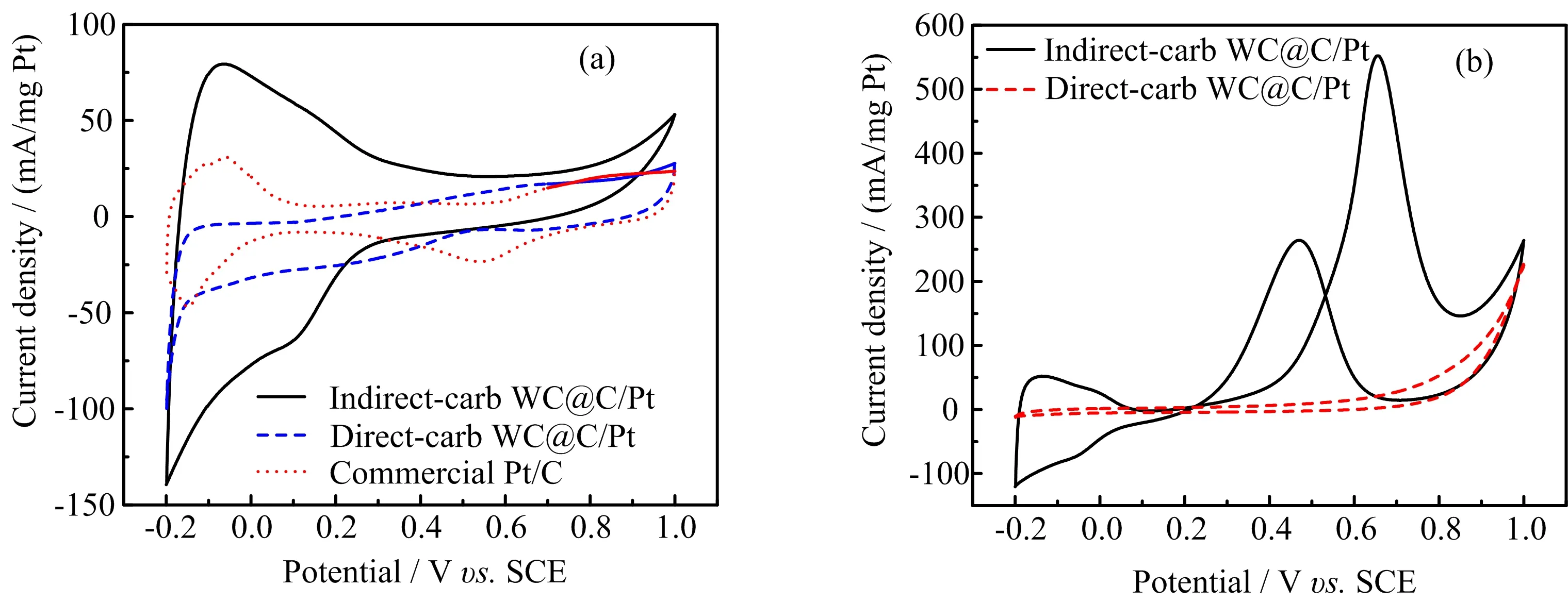
FIG.5(a)Cyclic voltammograms of indirect-carb WC@C/Pt,direct-carb WC@C/Pt and commercial Pt/C catalysts in N2-protecting 0.5 mol/L H2SO4electrolyte,(b)cyclic voltammograms curves on different electrodes in 0.5 mol/L H2SO4+0.5 mol/L CH3OH electrolyte at a scan rate of 50 mV/s.
The quantitative analysis of the elements from the STEM region of indirect-carb WC@C/Pt by EDX is shown in FIG.4(a),con firming the existence of W,C and Pt,which is also proven by the mapping images (FIG.3(d))and XRD curves(FIG.1(b)).However,the mass ratio of the WC to Pt will not be con firmed by the EDX.TG analysis under air was used to determine the percent WC present in indirect-carb WC@C/Pt sample. Assuming all carbon from WC and C was removed as CO2(with no other impurities presented),the mass of WC was calculated as follows:
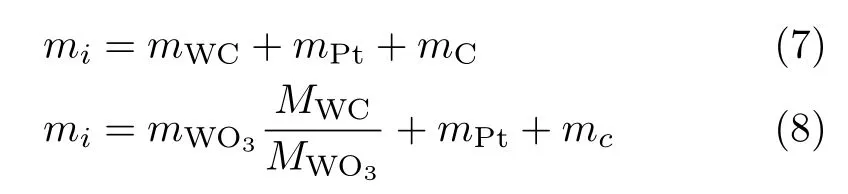
where miis initial mass,mWCis mass of pure WC,mPtis mass of Pt,mCis mass of carbon,mWO3is mass of pure WO3,MWCis molecular mass of WC,MWO3is molecular mass of WO3.
TG analyses of the WC samples under air show large mass gains of about 12.8 wt%(FIG.4(b),solid curve). WC and Pt were 69.6 wt%and 4.2 wt%(calculated by the Pt adding ratio vs.W)for indirect-carb WC@C/Pt sample,respectively.The oxidation point in air was also tested by the DTA result(FIG.4(b),broken cure). The sample would be oxidized and become stable during 420−580◦C.
The electrochemical measurements were used to characterize the surface composition of the prepared WC@C/Ptcatalyst. FIG.5(a)showstheCVs of indirect-carb WC@C/Pt,direct-carb WC@C/Pt, and commercial Pt/C electrodes in a N2-saturated 0.5 mol/L H2SO4solution. The comparison of the hydrogen adsorption/desorption behavior on different electrodes shows that the indirect-carb WC@C/Pt hybrid possesses a higher catalytic activity in the electro oxidation of hydrogen.The electrochemical surface area(ESA)of the Pt catalyst on the electrode surface has been estimated using the charge densities associated with adsorption/desorption of H de-rived from corresponding CV curves according to the reported literature[32]. The ESA of the indirectcarb WC@C/Pt(177.76 m2/g)is higher than commercial Pt/C(44.39 m2/g).However,the direct-carb WC@C/Pt shows no catalytic activity in the electro oxidation of hydrogen. The larger ESA of the indirect-WC@C/Pt catalyst can provide more active sites,which is signi ficant for enhanced electro activities of the indirect-WC@C/Pt catalyst.
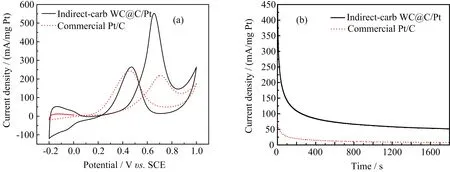
FIG.6(a)Cyclic voltammograms of indirect-carb WC@C/Pt and commercial Pt/C catalysts in N2-protecting 0.5 mol/L H2SO4+0.5 mol/L CH3OH electrolyte at a scan rate of 50 mV/s.(b)Chronoamperometry curves on different electrodes in 0.5 mol/L H2SO4+0.5 mol/L CH3OH electrolyte.
To assess their electro catalytic property in the methanol oxidations,the cyclic voltammograms curves on different electrodes were collected in 0.5 mol/L CH3OH containing 0.5 mol/L H2SO4solutions,as shown in FIG.5(b).The electro catalytic activities of WC@C/Pt catalysts towards methanol oxidations were found to be highly dependent on the microstructure in the composite.As presented in FIG.5(b),the indirectcarb WC@C/Pt catalysts displayed considerable activity for methanol oxidation,while there was no activity to the direct-carb WC@C/Pt catalysts.It could be ascribed to the latter lack of the electrochemical active sites due to the bigger particle size and aggregation of Pt on the surface of WC.
Additionally,the indirect-carb WC@C/Pt catalysts electro catalytic activity(552.16 mA/mg Pt)towards methanol oxidation was signi ficantly higher than that of Pt/C(218.93 mA/mg Pt),as could be seen from FIG.6(a).This was because Pt nanoparticles were uniformly dispersed in size/shape and randomly scattered across the in situ produced C by indirect carburization.In addition,in methanol electro oxidation,the tungsten carbide acts as adsorption site of hydroxyl ion,for which the intermediate species on Pt surface can be effectively removed on Pt surface during the methanol oxidation reaction.The larger ESA of the indirect-WC@C/Pt catalyst could provide more active sites,which was signi ficant for enhanced electro activities of the indirect-WC@C/Pt catalyst.Thus,we can expect that the highly improved electro catalytic properties of the indirect-WC@C/Pt may result from the above discussion.
To further compare the long-term electro catalytic stabilities of catalyst materials,the current densitytime curves are recorded in FIG.6(b).The indirectcarb WC@C/Pt and Pt/C catalysts present gradually decay of current density(55.49 mA/mg Pt vs.Pt/C 7.82 mA/mg Pt)with the extension of time due to gradual accumulation of intermediate products(CO)which poisons the catalysts.Besides,FIG.6(b)also shows that the indirect-carb WC@C/Pt catalyst possesses the best electrocatalytic stability of two catalysts due to the synergy between Pt and WC as discussed above.
IV.CONCLUSION
In summary,we have developed a simple effective method for the synthesis of discrete core-shell WC@C/Ptcatalyststhroughatypicalcarburization production mechanism at 800◦C.The spheres WC@C/Pt catalysts exhibited higher catalytic activity for electrochemical methanol oxidation reaction (552.16 mA/mg Pt vs.Pt/C 218.93 mA/mg Pt),and exceed 7 times higher in stability than a commercial Pt/C catalyst. By contrast,the as-prepared samples without the process of calcining showed no activity towards methanol oxidation due to lack of active sites.The thermal treatment promoted the formation of mesoporous structure and particles.It bene fited the Pt nanoparticles to be dispersed in size/shape and randomly scattered across the carbon spheres in the carbonizing process,which was believed to be the vital factor for the high quality catalysts.The current methods can be extended to synthesize other noble metals for wider ranges of electro catalysis and may find applications in a myriad of electrochemical processes.
V.ACKNOWLEDGMENTS
This work is supported by the International Science&Technology Cooperation Program of China(No.2010DFB63680),the Natural Science Foundation of Zhejiang Province(LQ15B030004)and Loughborough University and the EPSRC(EP/I013229/1).
[1]M.Winter and R.J.Brodd,Chem.Rev.104,42(2004).
[2]X.Yang,Q.D.Yang,J.Xu,and C.S.Lee,J.Mater. Chem.22,8057(2012).
[3]K.Mikkelsen,B.Cassidy,N.Hofstetter,L.Bergquist, A.Taylor,and D.A.Rider,Chem.Mater.26,6928 (2014).
[4]N.Zhang,S.J.Guo,X.Zhu,J.Guo,and X.Q.Huang, Chem.Mater.28,4447(2016).
[5]H.Zhang,M.S.Jin,and Y.N.Xia,Chem.Soc.Rev. 41,8035(2012).
[6]J.R.C.Salgado,F.Alcaide,G.Alvarez,L.Calvillo, and M.J.Lazaro,J.Power Sources 195,4022(2010).
[7]W.J.Zhou,Z.H.Zhou,S.Q.Song,W.Z.Li,G.Q. Sun,P.Tsiakaras,and Q.Xin,Appl.Catal.46,273 (2003).
[8]A.Chen and P.H.Hindle,Chem.Rev.110,3767 (2010).
[9]S.Bukola,B.Merzougui,A.Akinpelu,and M.Zeama, Electrochimica Acta 190,1113(2016).
[10]G.Singla,K.Singh,and O.P.Pandey,Int.J.Hydrogen Energy 40,5628(2015).
[11]Z.X.Yan,J.M.Xie,and P.K.Shen,J.Power Sources 286,239(2015).
[12]K.Woan,G.Pyrgiotakis,and W.Sigmund,Adv. Mater.21,2233(2009).
[13]S.Y.Zhang,N.P.Philipp,J.W.Joshua,S.Dai,M.J. Xu,G.W.George,C.Matteo,A.P.Frank,and X.Q. Pan,Nano Lett.16,4528(2016).
[14]R.B.Levy and M.Boudart,Science 181,547(1973).
[15]J.G.Chen,Chem.Rev.96,1477(1996).
[16]H.Chhina,S.Campbell,and O.Kesler,J.Power Sources 179,50(2007).
[17]R.H.Wang,C.G.Tian,L.Wang,B.L.Wang,H.B. Zhang,and H.G.Fu,Chem.Commun.3104(2009).
[18]G.S.Raman,J.H.Dong,and S.L.Jae,Electrochem. Commun.9,2576(2007).
[19]J.S.Moon,Y.W.Lee,S.B.Han,and K.W.Park,Int. J.Hydrogen Energy 39,7798(2014).
[20]R.Ganesan and J.S.Lee,Angew.Chem.Int.Ed.44, 6557(2005).
[21]C.A.Ma,Z.Y.Chen,W.F.Lin,F.M.Zhao,and M. Q.Shi,Micropor.Mesopor.Mater 149,76(2012).
[22]Y.Wang,S.Song,V.Maragou,P.K.Shen,and P. Tsiakaras,Appl.Catal.B 89,223(2009).
[23]Y.Wang,S.Q.Song,P.K.Shen,C.X.Guo,and C. M.Li,J.Mater.Chem.19,6149(2009).
[24]S.Shanmugam,D.S.Jacob,and A.Gedanken,J.Phys. Chem.B 109,19056(2005).
[25]V.G.Pol,S.V.Pol,and A.Gedanken,Adv.Mater. 23,1179(2011).
[26]V.G.Pol,S.V.Pol,and A.Gedanken,Adv.Mater. 18,2023(2006).
[27]F.P.Hu and P.K.Shen,J.Power Sources 173,877 (2007).
[28]Z.Y.Chen,C.A.Ma,Y.Q.Chu,J.M.Jin,X.Lin,C. Hardacre,and W.F.Lin,Chem.Commun.49,11677 (2013).
[29]G.J.French and F.R.Sale,J.Mater.Sci.16,3427 (1981).
[30]Z.Y.Chen,M.Q.Shi,C.A.Ma,Y.Q.Chu,and A. J.Zhu,Powder Technol.235,467(2013).
[31]J.Chen,B.Lim,E.P.Lee,and Y.Xia,Nano Today 4, 81(2009).
[32]Y.J.Li,W.Gao,L.J.Ci,C.M.Wang,and P.M. Ajayan,Carbon 48,1124(2010).
ceived on March 3,2017;Accepted on June 10,2017)
∗Authors to whom correspondence should be addressed.E-mail: chenzhy@zjut.edu.cn,chuyq@zjut.edu.cn
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Highly Responsive and Selective Ethanol Gas Sensor Based on Co3O4-Modi fied SnO2Nano fibers
- Intermolecular Interactions in Self-Assembly Process of Sodium Dodecyl Sulfate by Vertically Polarized Raman Spectra
- Identi fication of Superoxide O2−during Thermal Decomposition of Molten KNO3-NaNO2-NaNO3Salt by Electron Paramagnetic Resonance and UV-Vis Absorption Spectroscopy
- Binding Mechanism and Molecular Design of Benzimidazole/Benzothiazole Derivatives as Potent Abl T315I Mutant Inhibitors
- Geometric Design of Anode-Supported Micro-Tubular Solid Oxide Fuel Cells by Multiphysics Simulations
- Laser-Assisted Stark Deceleration of Polar Molecules HC2n+1N(n=2,3,4) in High-Field-Seeking State