Laser-Assisted Stark Deceleration of Polar Molecules HC2n+1N(n=2,3,4) in High-Field-Seeking State
2017-09-03KaiChenYunxiaHuangXiaohuaYang
Kai Chen,Yun-xia Huang,Xiao-hua Yang
School of Science,Nantong University,Nantong 226019,China
Laser-Assisted Stark Deceleration of Polar Molecules HC2n+1N(n=2,3,4) in High-Field-Seeking State
Kai Chen,Yun-xia Huang,Xiao-hua Yang∗
School of Science,Nantong University,Nantong 226019,China
Laser-assisted Stark deceleration scheme was proposed to decelerate the high- field-seeking molecule ICl in its rovibronic ground state.However,the laser intensity of 1.0×1010W/cm2is hard to realize in experiment.The time-of- flight signals of HC2n+1N(n=2,3 and 4) by three-dimensional Monte-Carlo simulation suggest that deceleration of such molecules is more feasible experimentally as only one-tenth laser intensity is needed.
Stark effect,Optical Stark effect,Molecular deceleration
I.INTRODUCTION
Molecules with rich internal structures provide the platform for studying the interaction dynamics of interand intra-molecules,and the interactions of molecules with external fields as well. However,the complex structures make us limit to such interactions accurately. Cold molecules by preparing molecules to low temperature region,due to clean quantum states and thus easier to be modeled,have made breakthroughs in the past two decades in this field,both experimentally and theoretically[1].Various methods have been carried out to obtain cold molecules,such as conformation of laser-cooled alkaline atoms[2,3],direct laser cooling of molecules[4,5],buffer gas cooling combined with bend guiding of polar molecules[6,7],Stark deceleration of polar molecules[8−12]and Zeeman deceleration of paramagnetic molecules[13−15].The anisotropic and long-range dipolar interactions add new ingredients to strongly correlated and collective quantum dynamics in many-body systems[16],so Stark deceleration of polar molecules is of great interest.Many weak- fieldseeking(WFS)molecules,CO[8],OH[10],H2CO[11], ND3[17],NH[12],SO2[18],YbF[19],and LiH[20] have been decelerated by employing this method to the equivalent temperature near 0.01 K so far.
However,most chemically stable(close shell)molecules are high- field-seeking(HFS)in their rovibronic ground states.Additionally,because the HFS molecules have no any scattering channel for inelastic collision [21],such cold molecules are probably cooled to ultralow temperature when trapped,and evaporatively cooled without any scattering loss by inelastic collision. Therefore,it is of particular importance to decelerate HFS molecules.A traditional Stark decelerator(SD) does not work for HFS molecules since such molecules will be transversely dispersed by the non-uniform electrostatic field produced by the deceleration stages[22]. Bethlem et al.[9]worked out an alternate gradient electrode array to decelerate HFS molecules,however,this scheme elongates the deceleration path greatly and the electrodes are difficult to be aligned experimentally[23].
Most recently,our group proposed a laser-assisted Stark deceleration(LSD)scheme[24]to decelerate ICl in its rovibronic ground state.We proposed that a far red-detuning laser beam,counter propagated to the molecular beam,can be used to guide HFS molecules transversely.In addition,the applied laser beam can also deepen the potential well in each stage,which will improve the deceleration efficiency(i.e.,lower velocity molecular package will be obtained).However,achieving the required laser intensity is experimentally challenging.To make it more feasible experimentally,linear HC2n+1N(n=2,3 and 4)molecules with large polarizability are recommended to be decelerated in the present work.Furthermore,the deceleration efficiency can be improved by amplitude-modulating the assisting laser.
II.APPARATUS AND THEORETICAL DESCRIPTION
The experimental apparatus of the switched-laserassisted Stark deceleration(SLSD)is similar to the LSD[24],as shown in FIG.1.The left part is a usual Stark decelerator merely omitting the hexapole(for focusing WFS molecules),which has been described in detail elsewhere[25],and the first deceleration stage locates 5 mm downstream the skimmer.The molecules HC2n+1N(n=2 and 3)can be synthesized by standard techniques of organic synthesis and HC9N can be prepared by mixed gas discharging[26,27].The sample molecules,HC2n+1N(n=2,3,and 4),buffered by xenon,at the backing pressure of about 1.0×105Paare supersonically expanded to a vacuum chamber via a liquid-nitrogen-cooled pulse valve.The molecular expansion then passes through a skimmer of 0.1 mm in diameter(matching the assisted laser beam)to form a molecular beam of a longitudinal central velocity of 280 m/s with 50 m/s full width at half magnitude and of a transverse temperature of about 10 mK.
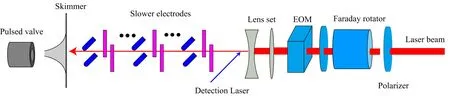
FIG.1 Experimental apparatus scheme of switched-laser-assisted Stark deceleration of polar molecules.
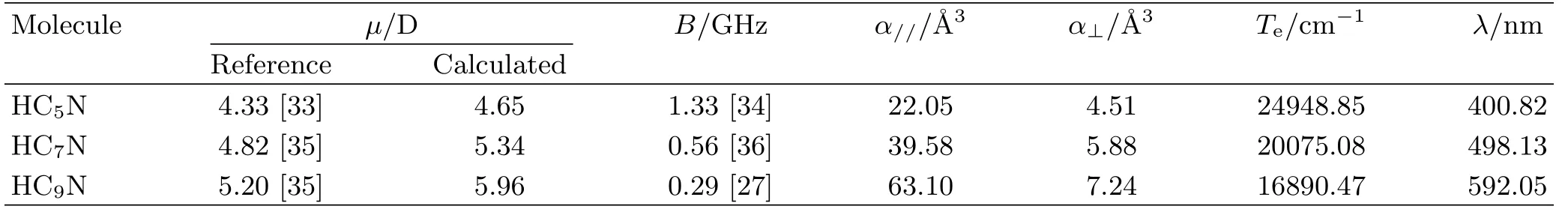
TABLE I Molecular parameters of HC2n+1N.
The molecular beam passes through the alternately perpendicular electrode pairs,which is identically spaced at 5.0 mm. Each electrode pair consists of two stainless steel rods,and±10 kV high voltages are applied to the pair to produce an electric field of 100 kV/cm at the center.A continuous wave far red-detuning laser beam,collimated by a lens set into 0.1 mm in diameter,co-axially but counter propagates against the molecular beam. The bene fits of the assisted-laser beam are:(i)to bunch molecular beam transversely due to optical Stark effect,and(ii) to give pseudo- first-order Stark effect to ground1Σ state molecules resulting in deeper potential well due to combined electrostatic and laser fields orientation[24]. Meanwhile,the linear polarization of the laser beam is modulated by an electro-optical modulator(EOM)to ensure that it is always parallel to the electric field experienced by the molecular package of interest.Before the laser beam enters EOM,it passes through a Faraday rotator.If the rotator switches the laser beam,it is SLSD,otherwise,it is simple LSD.The decelerated molecules can be detected after the slowing stages via either resonance enhanced multi-photon ionization or laser induced fluorescence spectroscopy.
In the rigid rotator model,the Hamiltonian of the molecule in combined electric and laser fields can be expressed as[28]:
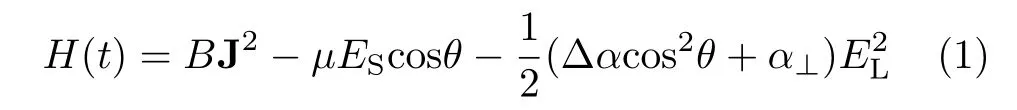
where B is molecular rotational constant,J is the molecular total angular momentum excluding nuclear spin,µis the molecular permanent electric dipole,θ is the angular of the molecular dipole with the electric field ES,the polarizability anisotropy∆α=α//−α⊥, where α//and α⊥are the polarizability components parallel and perpendicular to the molecular axis,respectively,and ESand ELare the amplitudes of electrostatic and laser fields.Therefore,the energy levels of the molecules in the combined fields can be obtained by diagonalizing the Hamiltonian matrix[29].
III.RESULTS AND DISCUSSION
Table I lists the molecular constants of HC2n+1N needed in the present work.Some constants are unavailable experimentally.Thus,we compute the values for the experimentally unknown constants employing the density functional theory(DFT).The geometry structures of the molecules are optimized under PBE1PBE/TZVP level[30,31],and the polarizability components are calculated under PBE1PBE/def2-TZVP level[31,32].To verify our prediction,our calculated values of permanent dipole moment are also listed in the second column ofµfor comparing with those of experiment or CCSD(T)theory( first column).Generally,the errors are within 15%,which implies that the DFT methodology works for our study.Note that,the values of the literatures(if available)are adopted in the following simulation.When calculating the electronic energy Teof the first excited state,the time-dependent DFT is employed to optimize the geometry structure under PBE1PBE/TZVP level[30,31].In Table I,λ is the wavelength of the resonance-exciting laser beam to excite the molecule into its first electronic excited state. Therefore,the wavelength of the detection laser is at λ, while the wavelength of the assisted laser beam is far red-detuning to λ.

TABLE II Values of∆E1,∆E2,∆E3and∆E4denoted in FIG.2.
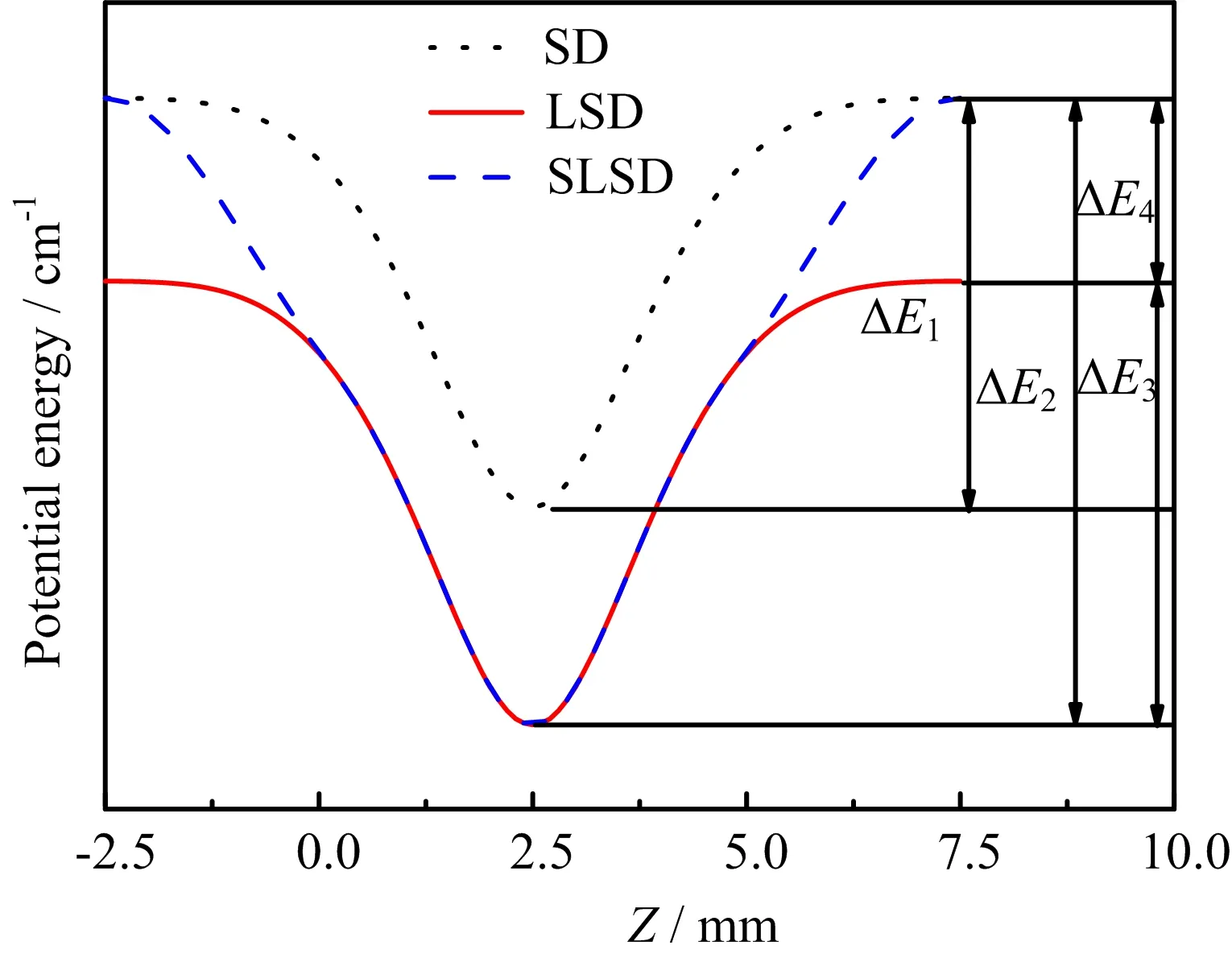
FIG.2 Spatial-dependentStark potentialcurvesof HC2n+1N in their high- field-seeking state|0,0〉.The dot line represents the potential in the usual Stark deceleration scheme,the solid line represents the potential in the laser-assisted Stark deceleration scheme,and the dash line represents the potential in the switched-laser-assisted Stark deceleration scheme with the laser switched o ffgradually at ϕ=0◦.
The potential of the|J,M〉=|0,0〉HFS state of HC2n+1N in the stage are plotted in FIG.2 and the values are listed in Table II,where the magnitude of the assisting laser intensity is set to be 1.0×109W/cm2. In FIG.2,the potential without any assisted laser field (SD),the potential with the laser field[24](LSD),and the potential with the switched-laser field(SLSD),are compared.Evidently,the molecule will lose up to∆E1kinetic energy when passing through each stage in SD, it will lose up to∆E3in LSD,and it will lose up to∆E2in SLSD.Therefore,the SLSD scheme is the most efficient for decelerating molecules.
Due to their lighter mass,larger permanent dipole moment and larger anisotropic polarizability of the HC2n+1N molecules compared to ICl,these molecules are experimentally more feasible to be decelerated to rest within 100 stages under the assisting laser intensity of 1.0×109W/cm2.In addition,as is evident in FIG.2,if switching the assisted laser beam,additional∆E4energy will be taken away when molecular beam passes through the electrode pairs.
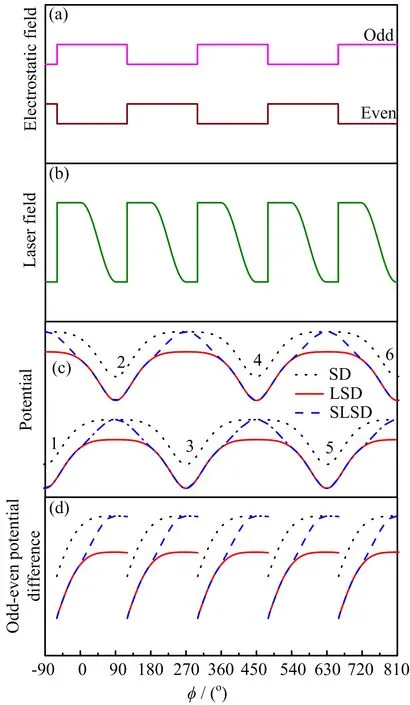
FIG.3 Switching time sequences of the odd and even electrostatic fields(a)and of the assisted laser field(b),and the potentials(c)molecules experienced(see FIG.2 for details) and the odd-even potential differences(d)when switching varying with ϕ.
The SD cannot decelerate molecules to rest due to the unavoidable low velocity loss[37],because the decelerating and bunching are achieved by the same electrodes.However,this kind of loss does not exist in LSD or SLSD because the decelerating and guiding are realized by the electrodes and laser beam,respectively.To compare the detail features of these three deceleration schemes,the control time sequences of the electrostatic field and the assisted laser are plotted in FIG.3,and the potentials and potential differences are plotted as well.The phase angle[37]is de fined as ϕ=zπ/L,where z is the longitudinal position originating from the middle of two successive stages and L is the spatial period of stages.In our Monte-Carlo simulation,a final velocity of 45 m/s and the laser intensity of 1.0×109W/cm2are chosen.
In addition,a time-of- flight(TOF)signals are plotted in FIG.4,where the down arrow indicates the TOF signal of the decelerated molecular package.FIG.4 tells that,it needs 52 and 53 stages to decelerate the HFS |0,0〉vibronic ground state of HC5N to the final velocity with the guiding efficiencies(de fined as the number ratio of the decelerated molecules to the incident molecules into the slower)of 0.30%and 0.39%in the SLSD and LSD respectively,while it needs 95 stages to decelerate the WFS|1,−1〉vibronic ground state of HC5N with the efficiency of 0.20%.The results are similar for HC7N and HC9N.Therefore,the SLSD and LSD are predicted to be more favorable than SD.
The one-tenth weaker laser intensity is still too intensive in experiment.To make the proposed scheme more feasible,we can choose a near-resonant red-detuning assisting laser,because the far red-detuning laser produces the AC Stark shift proportional to the laser intensity while the near-resonant red-detuning laser produces the AC Stark shift proportional almost to the square of the laser intensity[38].Modelling the nearresonant,red-detuning,optical Stark effect of molecules is in progress in our group.
IV.CONCLUSION
Decelerating HC2n+1N by both LSD and SLSD is studied in the present work.It is predicted that these HFS molecules can be decelerated to rest utilizing less than 100 stages.That the assisted laser intensity can be one-tenth weak of the previous proposal of the deceleration of ICl[24]makes it more feasible experimentally.Prospectively,near-resonant red-detuning laser beam employed as the assisted laser will be much more experimentally practical.
V.ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China(No.20273066).
[1]S.Y.T.van de Meerakker,H.L.Bethlem,N.Vanhaecke,and G.Meijer,Chem.Rev.112,4828(2012).
[2]A Vardi,M.Shapiro,and K.Bergmann,Opt.Express 4,91(1999).
[3]K.K.Ni,S.Ospelkaus,M.H.G.de Miranda,A.Pe’Er, B.Neyenhuis,J.J.Zirbel,S.Kotochigova,P.S.Julienne,D.S.Jin,and J.Ye,Science 322,231(2008).
[4]M.T.Hummon,M.Yeo,B.K.Stuhl,A.L.Collopy,Y. Xia,and J.Ye,Phys.Rev.Lett.110,143001(2013).
[5]J.F.Barry,E.S.Shuman,E.B.Norrgard,and D. Demille,Phys.Rev.Lett.108,103002(2012).
[6]S.A.Rangwala,T.Junglen,T.Rieger,P.W.H.Pinkse, and G.Rempe,Phys.Rev.A 67,043406(2002).
[7]T.Junglen,T.Rieger,S.A.Rangwala,P.W.H.Pinkse, and G.Rempe,Phys.Rev.Lett.92,223001(2004).
[8]H.L.Bethlem,G.Berden,and G.Meijer,Phys.Rev. Lett.83,1558(1999).
[9]H.L.Bethlem,F.M.H.Crompvoets,R.T.Jongma, S.Y.T.van de Meerakker,and G.J.M.Meijer,Phys. Rev.A 65,053416(2002).
[10]J.R.Bochinski,E.R.Hudson,H.J.Lewandowski,G. Meijer,and J.Ye,Phys.Rev.Lett.91,243001(2003).
[11]E.R.Hudson,C.C.Ticknor,B.C.Sawyer,C.A.Taatjes,H.J.Lewandowski,J.R.Bochinski,J.L.Bohn,and J.Ye,Phys.Rev.A 73,063404(2005).
[12]S.Y.T.van de Meerakker,I.Labazan,S.Hoekstra,J. Kpper,and G.Meijer,J.Phys.B:At.Mol.Opt.Phys. 39,S1077(2005).
[13]N.Vanhaecke,U.Meier,M.Andrist,B.H.Meier,and F.Merkt,Phys.Rev.A 75,031402(2007).
[14]S.D.Hogan,A.W.Wiederkehr,H.Schmutz,and F. Merkt,Phys.Rev.Lett.101,143001(2008).
[15]E.Narevicius,A.Libson,C.G.Parthey,I.Chavez,J. Narevicius,U.Even,and M.G.Raizen,Phys.Rev.A 77,051401(2008).
[16]M.A.Baranov,A.Micheli,S.Ronen,and P.Zoller, Physics 83,043602(2010).
[17]H.L.Bethlem,G.Berden,F.M.H.Crompvoets,R.T. Jongma,A.J.A.van Roij,and G.Meijer,Nature 406, 491(2000).
[18]S.Jung,E.Tiemann,and C.Lisdat,Phys.Rev.A 74, 040701(2006).
[19]M.R.Tarbutt,H.L.Bethlem,J.J.Hudson,V.L. Ryabov,V.A.Ryzhov,B.E.Sauer,G.Meijer,and E. A.Hinds,Phys.Rev.Lett.92,173002(2004).
[20]S.K.Tokunaga,J.O.Stack,J.J.Hudson,B.E.Sauer, E.A.Hinds,and M.R.Tarbutt,J.Chem.Phys.126, 124314(2007).
[21]J.L.Bohn,Phys.Rev.A 63,052714(2001).
[22]H.L.Bethlem,A.J.A.van Roij,R.T.Jongma,and G.Meijer,Phys.Rev.Lett.88,133003(2002).
[23]T.E.Wall,J.F.Kanem,J.M.Dyne,J.J.Hudson,B. E.Sauer,E.A.Hinds,and M.R.Tarbutt,Phys.Chem. Chem.Phys.13,18991(2011).
[24]Y.X.Huang,S.W.Xu,and X.H.Yang,J.Phys.B: At.Mol.Opt.Phys.49,135101(2016).
[25]F.M.H.Crompvoets,R.T.Jongma,H.L.Bethlem, A.J.A.van Roij,and G.Meijer,Phys.Rev.Lett.89, 093004(2002).
[26]M.C.McCarthy,M.J.Travers,A.Kovcs,C.A.Gottlieb,and P.Thaddeus,Astrophys.J.Suppl.Ser.113, 105(1997).
[27]M.Iida,Y.Ohshima,and Y.Endo,Astrophys.J.371, L45(1991).
[28]B.Friedrich and D.Herschbach,J.Phys.Chem.A 103, 10280(1999).
[29]M.H¨artelt and B.Friedrich,J.Chem.Phys.128, 224313(2008).
[30]J.P.Perdew,K.Burke,and M.Ernzerhof,Phys.Rev. Lett.77,3865(1996).
[31]A.Sch¨afer,C.Huber,and R.Ahlrichs,J.Chem.Phys. 100,5829(1994).
[32]F.Weigend and R.Ahlrichs,Phys.Chem.Chem.Phys. 7,3297(2005).
[33]H.S.Liszt,J.Pety,and R.Lucas,Astron.Astrophys. 486,493(2008).
[34]L.W.Avery,T.Oka,N.W.Broten,and J.M. MacLeod,Astrophys.J.231,48(1979).
[35]P.Botschwina and M.Horn,J.Mol.Spectrosc.185, 191(1997).
[36]H.W.Kroto,C.Kirby,D.R.M.Walton,L.W.Avery, N.W.Broten,J.M.Macleod,and T.Oka,Astrophys. J.219,L133(1978).
[37]E.R.Hudson,J.R.Bochinski,H.J.Lewandowski,B. C.Sawyer,and J.Ye,Eur.Phys.J.D 31,351(2004).
[38]H.Stapelfeldt and T.Seideman,Rev.Mod.Phys.75, 543(2003).
ceived on April 24,2017;Accepted on June 3,2017)
∗Author to whom correspondence should be addressed.E-mail: xhyang@ntu.edu.cn
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- Highly Responsive and Selective Ethanol Gas Sensor Based on Co3O4-Modi fied SnO2Nano fibers
- Intermolecular Interactions in Self-Assembly Process of Sodium Dodecyl Sulfate by Vertically Polarized Raman Spectra
- Identi fication of Superoxide O2−during Thermal Decomposition of Molten KNO3-NaNO2-NaNO3Salt by Electron Paramagnetic Resonance and UV-Vis Absorption Spectroscopy
- Binding Mechanism and Molecular Design of Benzimidazole/Benzothiazole Derivatives as Potent Abl T315I Mutant Inhibitors
- Fabricating Core-Shell WC@C/Pt Structures and its Enhanced Performance for Methanol Electrooxidation
- Geometric Design of Anode-Supported Micro-Tubular Solid Oxide Fuel Cells by Multiphysics Simulations