Binding Mechanism and Molecular Design of Benzimidazole/Benzothiazole Derivatives as Potent Abl T315I Mutant Inhibitors
2017-09-03WeicongLinShepeiTnShengfuZhouXiojieZhengWenjunWuKngchengZheng
Wei-cong Lin,She-pei Tn,Sheng-fu Zhou,Xio-jie Zheng,Wen-jun Wu∗,Kng-cheng Zheng
a.Department of Physical Chemistry,Guangdong Pharmaceutical University,Guangzhou 510006,China
b.School of Chemistry and Chemical Engineering,Sun Yat-sen University,Guangzhou 510275,China
Binding Mechanism and Molecular Design of Benzimidazole/Benzothiazole Derivatives as Potent Abl T315I Mutant Inhibitors
Wei-cong Lina,She-pei Tana,Sheng-fu Zhoua,Xiao-jie Zhenga,Wen-juan Wua∗,Kang-cheng Zhengb
a.Department of Physical Chemistry,Guangdong Pharmaceutical University,Guangzhou 510006,China
b.School of Chemistry and Chemical Engineering,Sun Yat-sen University,Guangzhou 510275,China
Despite the efficacy of imatinib therapy in chronic myelogenous leukemia,the development of drug-resistant Abl mutants,especially the most difficult overcoming T315I mutant,makes the search for new Abl T315I inhibitors a very interesting challenge in medicinal chemistry.In this work,a multistep computational framework combining the three dimensional quantitative structure-activity relationship(3D-QSAR),molecular docking,molecular dynamics(MD)simulation and binding free energy calculation,was performed to explore the structural requirements for the Abl T315I activities of benzimidazole/benzothiazole derivatives and the binding mechanism between the inhibitors and Abl T315I.The established 3D-QSAR models exhibited satisfactory internal and external predictability.Docking study elucidated the comformations of compounds and the key amino acid residues at the binding pocket,which were con firmed by MD simulation.The binding free energies correlated well with the experimental activities.The MM-GBSA energy decomposition revealed that the van der Waals interaction was the major driving force for the interaction between the ligands and Abl T315I.The hydrogen bond interactions between the inhibitors and Met318 also played an important role in stablizing the binding of compounds to Abl T315I.Finally, four new compounds with rather high Abl T315I activities were designed and presented to experimenters for reference.
Abl T315I mutant inhibitor,Benzimidazole/benzothiazole derivative,Three dimensional quantitative structure-activity relationship,Docking study,Molecular dynamics simulation,Molecular design
I.INTRODUCTION
Bcr-Abl is a constitutively activated cytoplasmic tyrosine kinase(TK)encoded by the Philadelphia chromosome,which derives from the reciprocal translocation between chromosomes 9(Abelson oncogene,Abl) and 22(breakpoint cluster region gene,Bcr).This recombined gene produces the fusion protein that drives clonal malignancy characterized by unchecked myeloid proliferation[1,2].The finding that Bcr-Abl is the etiologic agent of chronic myeloid Leukemia(CML),being present in most cases of CML,and that the Bcr-Abl activity is fundamental Bcr-Abl-mediated transformation makes this kinase an important target for the development of speci fic therapies,especially for the treatment of CML.Imatinib is a potent first-generation Bcr-Abl kinase inhibitor approved for clinical use and effective for most CML patients.Although clinical remission has been achieved with imatinib,many patients relapse because of the point mutations in the Bcr-Abl kinase domain.So far,more than 100 different point mutations have been reported in patients with imatinib-resistant CML,among which the T315I mutation is of particular concern.T315I mutation is the most difficult to be overcome and to date less compounds can inhibit cases with T3151 mutation that accounts for about 15%−20% of all clinically relevant CML mutations[3].Because this mutation occurs at the gatekeeper site and may cause steric hindrance precluding the access of the inhibitors to ATP-binding pocket[4],the discovery of potent Bcr-Abl inhibitors targeting T315I mutation would have important implications to treat the most clinically promising Abl T315I mutant therapies.Several new compounds,such as ponatinib[5]and rebastinib[6], have been studied clinically for CML patients harboring the Bcr-Abl T315I mutation,and it has shown that the Bcr-Abl inhibitors targeting the T315I mutant can inhibit the full range of Bcr-Abl kinase domain mutations as well as the wild-type kinase[7].
Recently,Hong and co-workers reported that benzimidazole/benzothiazole is a high-affinity inhibitor targeting T315I mutant Bcr-Abl to treat CML and have synthesized a novel series of benzimidazole/benzothiazolederivatives[8].They assessed their activities and discovered that these compounds possessed remarkable Bcr-Abl inhibitory activity at low nanomolar concentrations and potently inhibit several human cancer cell lines,demonstrating the great potential for developing these compounds as a novel class of Bcr-Abl inhibitors for T315I mutant therapy.Although some progresses have been made in experimental researches,so far the theoretical studies on the mechanisms of these compounds toward Bcr-Abl kinase and the structural features in fluencing their antileukemia activities remain largely unknown.
To predict the drug discovery process,it is important to perform the theoretical studies on the mechanisms of ligand-receptor interactions in detail.3DQSAR studies,including comparative molecular field analysis(CoMFA)and comparative molecular similarity indexes analysis(CoMSIA)have been successfully used in understanding the pharmacological properties of the studied compounds and modern drug design,not only because the 3D-QSAR models give a visual output but also the obtained field maps may help to understand the detailed 3D-structure of the active site of a receptor[9,10].Molecular docking is also a helpful methodology to further study the interaction mechanisms,as it can simulate the vivid interaction picture between a ligand and a receptor[11].In addition,molecular dynamics simulation studies the interaction mechanism of protein complex with ligands by offering vivid pictures to depict the fluctuations and conformational changes of molecules,and the structural features contributing to the inhibitory activity[12].Therefore,a combined 3D-QSAR,molecular docking and MD simulation study can provide deep insights into the understanding structural characteristics of ligand-Abl interactions and the binding process.
In this study, a novelseries ofbenzimidazole/benzothiazole derivatives as Abl T315I mutant inhibitors were selected to perform a multistep computational framework by using molecular dockings,3DQSAR,MD simulations and binding free energy calculations.The rational binding modes and detailed interactions of these compounds with Abl T315I kinase were analyzed,and the optimum 3D-QSAR models were established.In particular,some key structural features and key residues responsible for the inhibition toward Abl T315I were also revealed,and four new compounds with higher Abl T315I inhibitory activity were designed.We expect the obtained results can offer some useful references for the experimental work.
II.COMPUTATIONAL METHODS
A.Data set
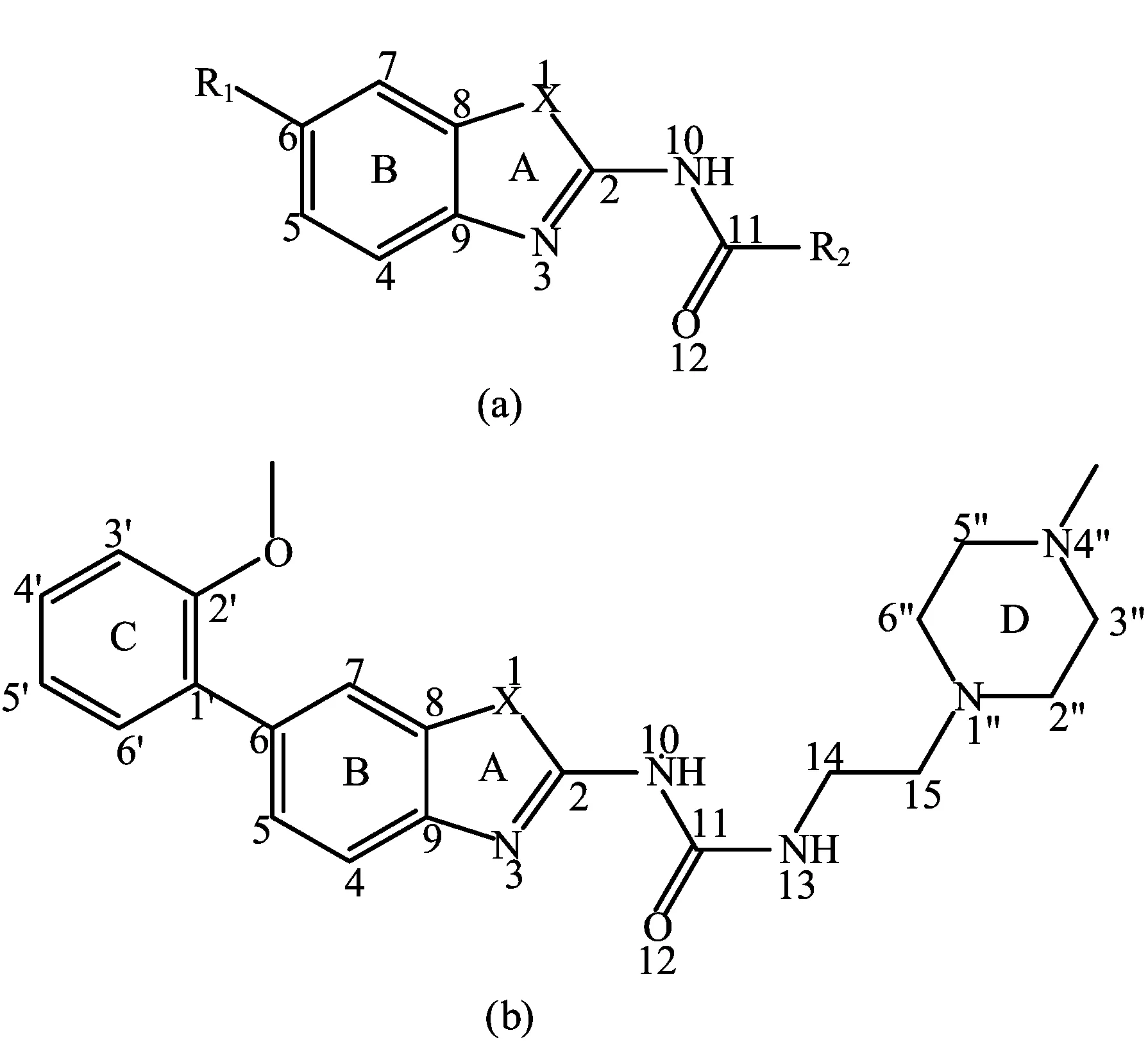
FIG.1 General structural formula and numbering of(a)the studied nocodazole derivatives and(b)template molecule compound 42.
To perform this study,a set of 48 variously functionalized benzimidazole/benzothiazole derivatives exhibiting potent inhibitory activity against Abl T315I kinase were obtained from published literature[8].The general structural formula of the studied compounds are shown in FIG.1.The total set of these derivatives was divided into a training set(34 compounds)to generate the 3D-QSAR models and a test set(14 compounds)to evaluate the predictive ability of the developed models (Table I).The test compounds were selected manually considering the structural diversity and wide range of activities in the data set.All original IC50values were converted to pIC50values and used as dependent variables in the 3D-QSAR analyses.
The 3D-structures of the benzimidazole/benzothiazole derivatives were constructed by the sketch Molecule module in Sybyl 6.9 software.Structural energy minimization was performed using Powell gradient algorithm and the Tripos force field with a convergence criterion of 0.001 kcal/(mol·˚A)and a maximum of 1000 iterations.MMFF94 charges were assigned to each compound.The minimized structure was used as the initial conformation for molecular docking.
B.Molecular docking
To acquire the probable binding conformations and orientations of the studied compounds interacting with Abl T315I kinase,docking studies were carried out with the sur flex module in Sybyl 6.9 software package[13].
The X-ray crystal structure of Abl T315I taken from the Protein Data Bank(PDB ID 2Z60)was used to dock. At the beginning of docking,all the water molecules and subunits were removed,polar hydrogen atoms were added and Kollman all atom charges were assigned to the protein.In this study,the ligand-based mode was adopted to generate the protomol,with twoimportant parameters,i.e.,protomol threshold and protomol bloat at their default values of 0.5 and 0,respectively.With the other default parameters used,20 conformations per ligand were acquired by Sur flex-dock method,and the binding conformation with the highest docking score and its orientation being similar to that of the cocrystallized ligand was selected for further 3DQSAR studies.In the molecular docking,the inhibitors were regarded as being flexible and the protein was considered to be rigid.
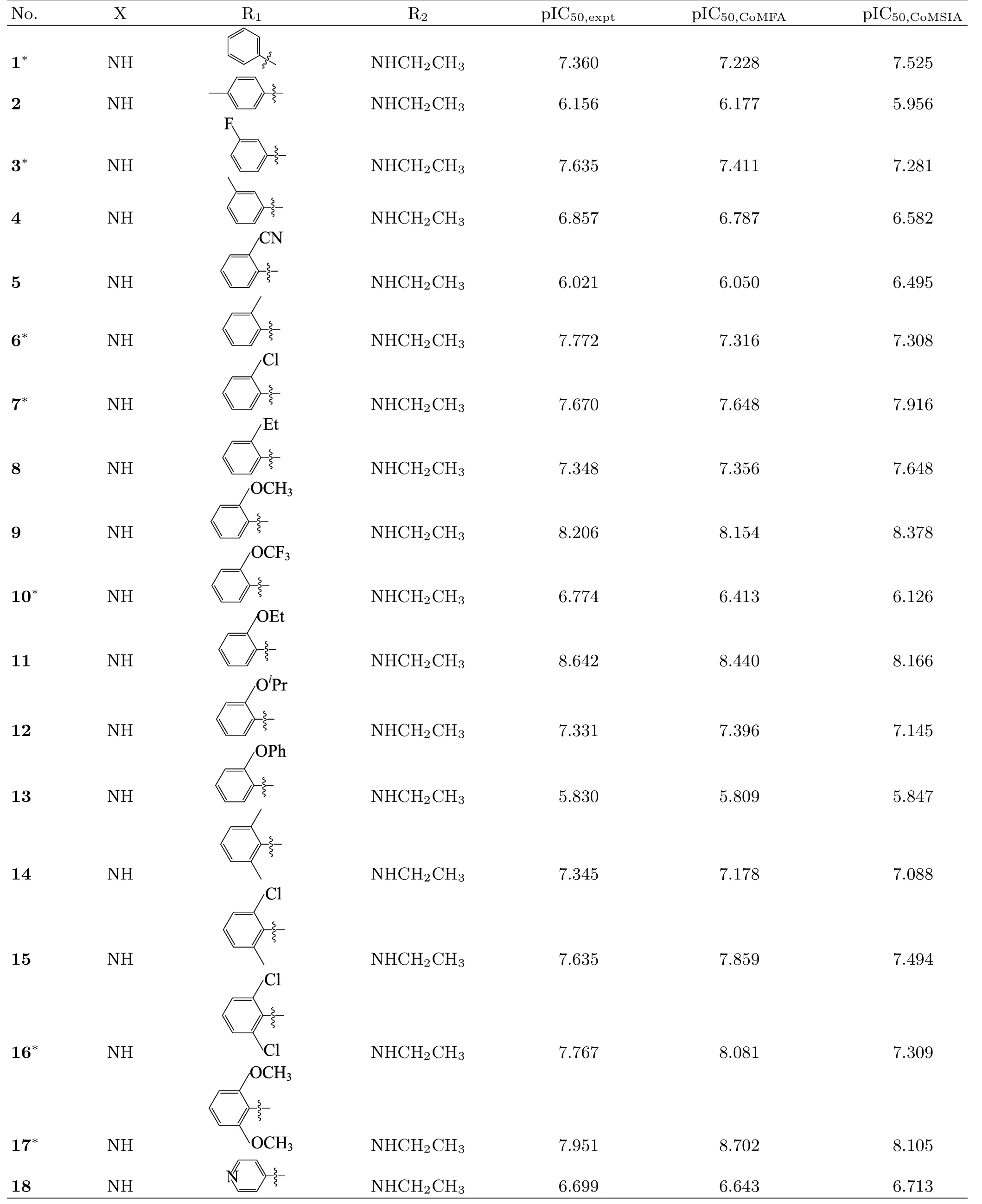
TABLE I Structures and 3D-QSAR results of the studied compounds.
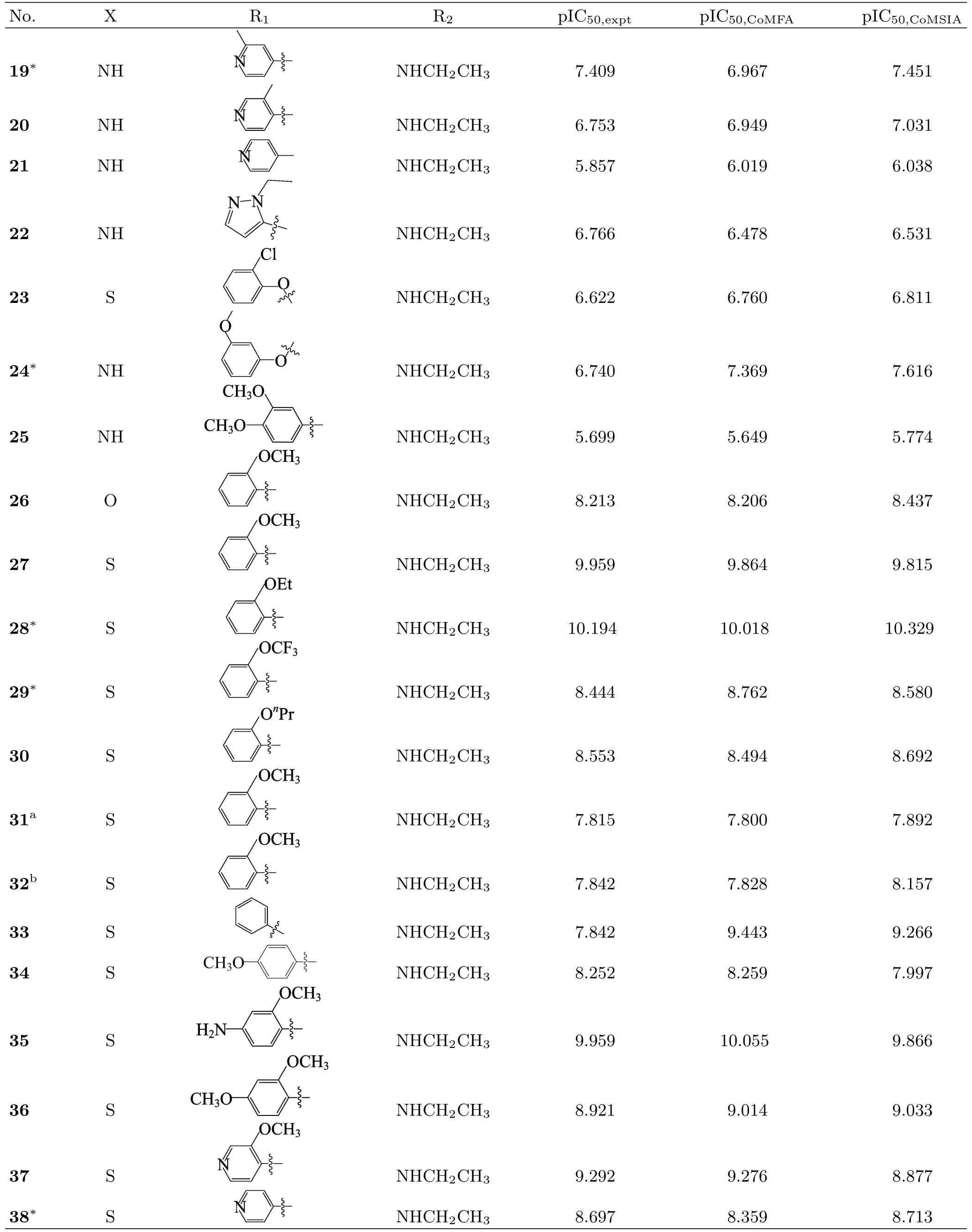
Table I Continued.
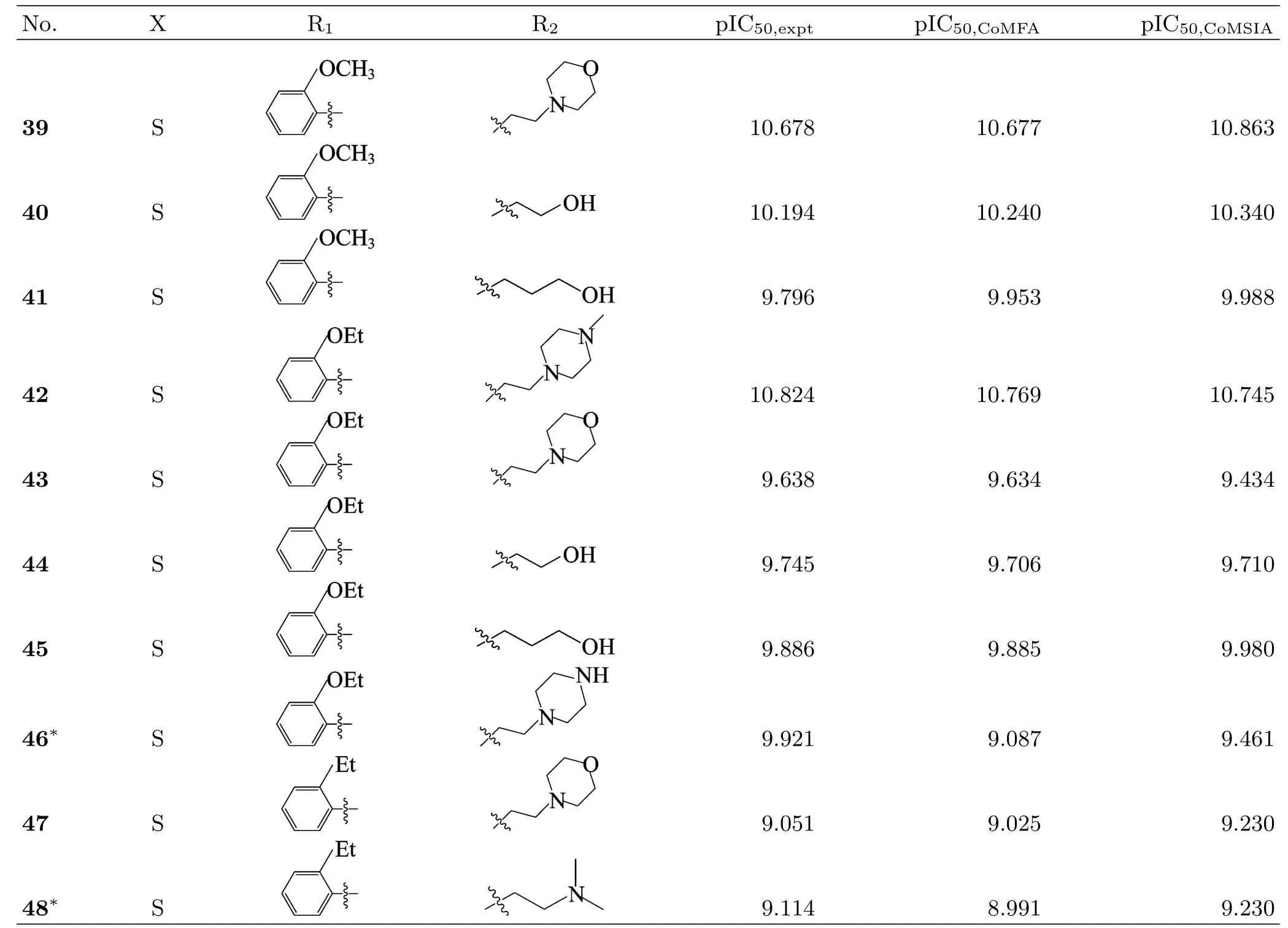
Table I Continued.
C.Molecular modeling and alignment
CoMFA and CoMSIA studies were performed by using SYBYL 6.9 molecular modeling software package running on an SGI R2400 workstation.All parameters used in COMFA and CoMSIA were default except for explaination.
Active conformation selection and structural alignment of compounds are key steps for 3D-QSAR analysis[14,15].Two different alignment methods were applied to obtain the optimal 3D-QSAR models.One is the ligand-based alignment,i.e.,all compounds were aligned to the most potent compound 42 resulted by the Align Database command in SYBYL 6.9 software.The other one is the receptor-based alignment,in which the optimal conformations of all compounds derived from docking studies were assigned to MMFF94 charges and imported into molecular alignment for 3D-QSAR analysis.
D.Generation of 3D-QSAR model
In CoMFA analysis,models of steric and electrostatic fields were based on both Lennard-Jones and Coulombic potentials.Steric and electrostatic energies were calculated using Tripos force field with a distance dependent dielectric constant at all intersections in a regularspaced(2˚A)grid taking an sp3carbon atom as steric probe and a+1 charge as electrostatic probe.The truncation for both the steric and the electrostatic energies was set to 30 kcal/mol[16,17].
In CoMSIA, five molecular fields were calculated: steric, electrostatic, hydrophobic, hydrogen bond donor,and hydrogen bond acceptor.The CoMSIA descriptors were derived by using a 3D cubic lattice with grid spacing of 2˚A and extending 4˚A units beyond the aligned molecules in all directions.A sp3carbon probe atom with a charge of+1.0,a van der Waals radius of 1.0˚A,hydrophobicity of+1.0,and H-bond donor and acceptor properties of+1 were used to calculate the corresponding fields.A Gaussian-type functional form was adopted to evaluate the mutual distance between the probe atom and each molecule atom.The attenuation factor was set to the default value of 0.3.
E.Partial least squares analysis and validation of QSAR models
The 3D-QSAR equations were generated using the partial least-square(PLS)statistical method.PLS algorithm with the leave-one-out(LOO)cross-validation method was performed to determine the highest crossvalidation correlation coefficient(q2)and the optimum number of components N.Then,the non-crossvalidation analysis was applied to calculate the conventional correlation coefficient R2,standard error of estimates(SEE),and F value.To further assess the robustness and statistical validity of the derived models, bootstrapping analysis[18]for 100 runs was also carried out.
To assess the predictive abilities of the 3D-QSAR models obtained from the training set,the pIC50values of 14 compounds in the external test set were predicted. The predictive power of the models is judged,depending on the predictive correlation coefficient(R2pred)calculated by the following formula:

where PRESS is the sum of squared deviations between the experimental and predicted activities of the test set compounds,and SD denotes the sum of squared deviation between the biological activities of the training set molecules and the mean activity of the training set molecules.
F.Molecular dynamics(MD)simulations
Based on docking results,MD simulations were carried out with AMBER 9 software package[19,20].The docked structures of 2Z60 with two compounds(the most active compound 42 and least active compound 25)were used as the initial structures for MD calcutions.The electrostatic potentials(ESP)of the ligands were calculated at the B3LYP/6-31G(d)level in the Gaussian 09 program,and the partial atomic charges for ligand atoms were generated by using the RESP method implemented in the Antechamber module.The FF03 AMBER force field and the general AMBER force field(ga ff)[21]were respectively used to describe the protein and the ligands.The complexes were neutralized by adding sodium ions and solvated in a truncated octahedral box of TIP3P water model[22]and the distance between the solute surface and the edge of thebox was chosen to be larger than 12
Before the MD simulations,energy minimizations were performed to relax the complex systems using two stages.In first stage,the complex was constrained with a restraint constant of 2.0 kcal/(mol·˚A2)and the waters and sodium ions were energetically minimized by 2000 steps of steepest descent method followed by 3000 steps of conjugated gradient method.In second stage,the whole relaxed complex was optimized by 5000 steepest descent minimization and 5000 steps conjugate gradient minimization.Afterward,the systems were gradually heated from 0 to 300 K in 200 ps with a force constant of 1.0 kcal/(mol·˚A2),and equilibrated for 500 ps at 300 K and 1 atm.Finally,12.0 ns production MD simulation was carried out in the NPT ensemble at 300 K with 1.0 atm pressure.During the simulations,the Particle Mesh Ewald(PME)method[23]was used to treat the long-range electrostatic interactions with the nonbonded cuto ffof 8˚A,and SHAKE algorithm[24]was applied to restrain all covalent bonds including hydrogen atoms with 2 fs time step.The coordinated trajectories files of production MD simulation were stored every 1 ps and the stability of the complexes was veri fied by the root mean square deviation(RMSD).500 snapshots of the simulated conformation extracted from the last 2 ns MD trajectories were used to perform the following binding free energy calculations.
G.Binding free energy calculations
To verify the binding stability of the ligand-protein complexes,the free energy calculations were performed by the molecular mechanics Poisson-Boltzmann surface area(MM-PBSA)approach[25].For each snapshot,the free energy was calculated for each molecular species (complex,receptor and ligand),and the binding free energy is calculated by the following equation:
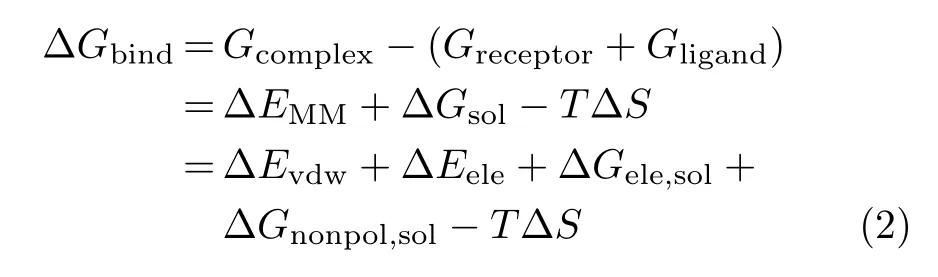
where∆EMMrepresents the molecular mechanical free energy(including van der Waals interaction energy∆Evdwand electrostatic interaction energy∆Eele).∆Gsolis de fined as the solvation free energy(consisting of the polar solvation free energy∆Gele,soland the nonpolar solvation free energy∆Gnonpol,sol).The∆Gele,solwas evaluated by the generalized Born(GB)approximation model,while the∆Gnonpol,solwas calculated using the solvent accessible surface area(SASA)that was determined by the Molsurf module in AMBER9.0.T∆S is the entropy contribution which was omitted in this study due to its relatively high computational demand in Amber 9.0 and the slight variation of substitution in the two inhibitors.
To obtain the detailed interactions between protein and ligand,the binding free energies were decomposed to single residues using MM-GBSA method.The contribition of each residue includes four energy terms: an electrostatic interaction energy(∆Gele),a van der Waals interaction energy(∆Gvdw),a polar solvation free energy(∆Gele,sol)and the nonpolar solvation free energy(∆Gnonpol,sol).
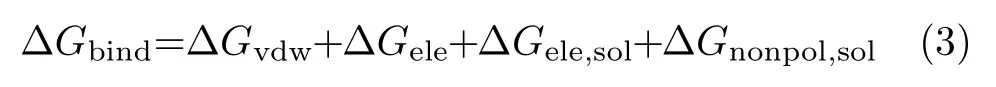
where the∆Gvdwand∆Gelewere determined by sander program in Amber 9.0.The polar contribution was computed with the generalized Born model (GBOBC,igb=2)[26]. The nonpolar contribution was calculated using the solvent accessible surface area (SASA)[27,28].
III.RESULTS AND DISCUSSION
A.Docking results
Before docking,it was important to validate the reliability of the docking project.The native ligand P3Y was extracted from the X-ray structure of Abl T315I (PDB entry:2Z60)and redocked to the protein binding pocket.As a result,the root-mean-square deviation (RMSD)value between the conformation of docked P3Y and crystallographic P3Y was 0.68˚A,suggesting a high reliability of docking procedure.
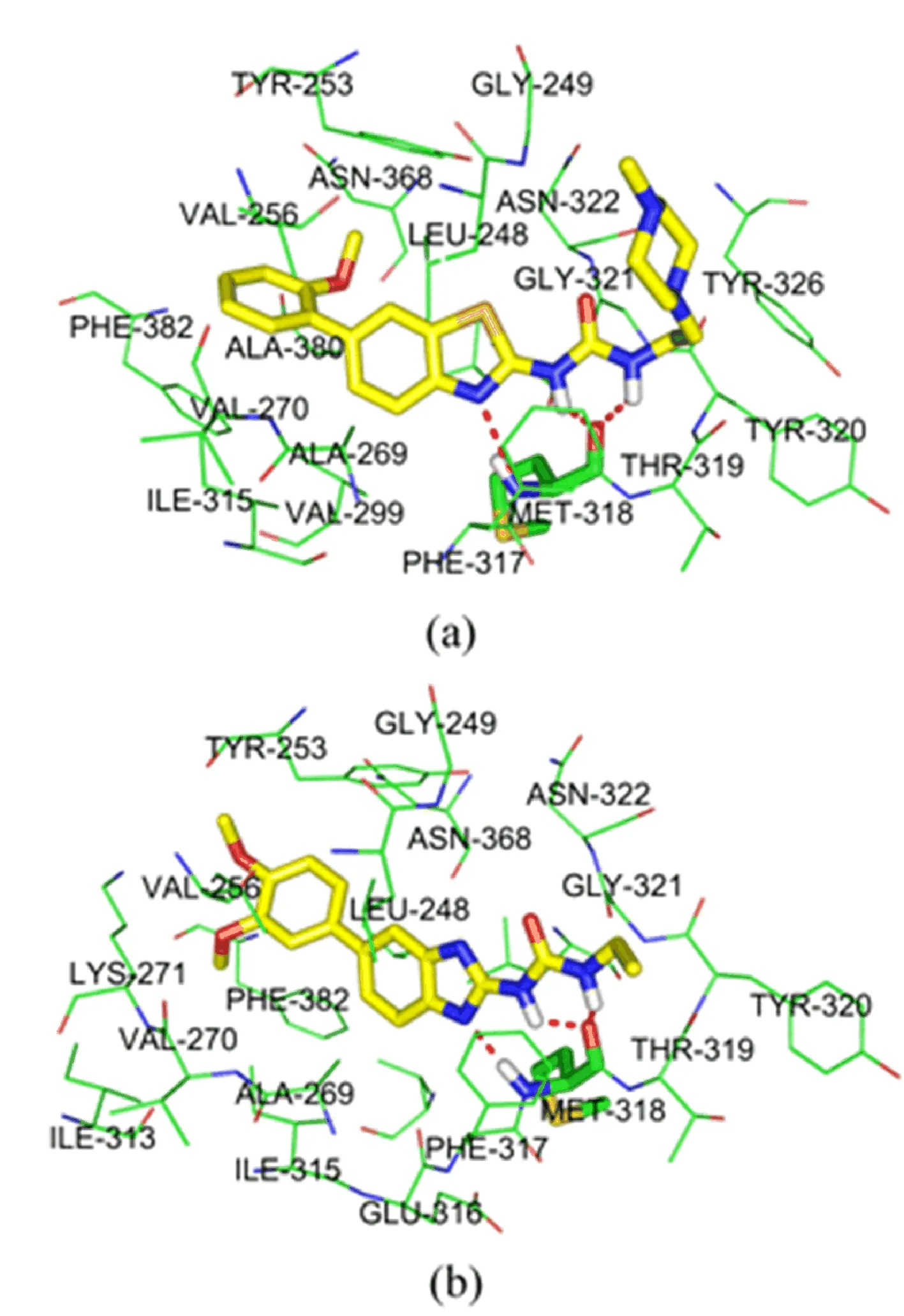
FIG.2 Docking structure of(a)the most potent compound 42 and(b)the least potent compound 25.The dashed line represents a hydrogen bond between ligands and protein.
In this work,all 48 studied inhibitors were successfully docked into the binding pocket of Abl T315I.In order to further elucidate the interaction mechanism, the most active compound 42 and the least active compound 25 were selected to perform the deeper docking analysis and their binding models with Abl T315I kinase were displayed in FIG.2.It can be seen that two compounds are suitably located at the ATP binding site of Abl T315I and result in various interactions with the active regions of the enzyme.They can all form three hydrogen bonds with the hinge region:one between the N3 of the thiazole(or imidazole)ring and the NH of Met318,the other two between the hydrogens of N10H and N13H groups and the carbonyl oxygen of Met318, with corresponding bond lengths of 3.2,1.7,and 2.4˚A for 42,and 3.1,2.0,and 2.2˚A for 25,respectively.Except for the hydrogen bond interactions,the hydrophobic interaction should also be taken into consideration to explain the activity difference of two compounds. As can be seen in FIG.2,the substituent R1of two compounds penetrates deeply into a large hydrophobic pocket,in van der Waals contact with Leu248,Tyr253, Val256,Ala269,Val270,Ala380,and Phe382.Because the hydrophilic nature of 3,4-dimethoxyphenyl of 25 gets stronger than that of 2-methoxyphenyl of 42,we can infer that compound 42 might form stronger hydrophobic interactions with Abl T315I than compound 25. And the terminal two methoxy groups of substituent R1in 25 are blocked by the side chains of Val256 and Phe382,due to its positional shift away from the ATP-binding site to the solvent region.This strengthening of repulsive interactions,resulting in the less stablization in the ATP-binding site of terminal 3,4-dimethoxyphenyl of 25 than 2-methoxyphenyl of 42, may be an explanation for the signi ficant decrease in the inhibitory activity from 42 to 25.At the same time,the 4-methylpiperazine of substituent R2in 42 is oriented toward the solvent region,and easily forms favorable interactions with plasma protein.While the ethylamine group of 25 is buried completely in a small pocket created by Met318,Thr319,Tyr320,and Gly321.This favorable interactions with plasma protein are also ob-viously responsible for the inhibitory activity of 42 than 25.In addition,the thiazole ring of 42(or imidazole ring of 25)may form the π-π stacking with Phe317. Therefore,all of the above analyses conformably show that the hydrophobic interaction,repulsive interaction, as well as the solvent interaction may be the important factors to elucidate the differences in inhibitory activities of compound 42 and 25.
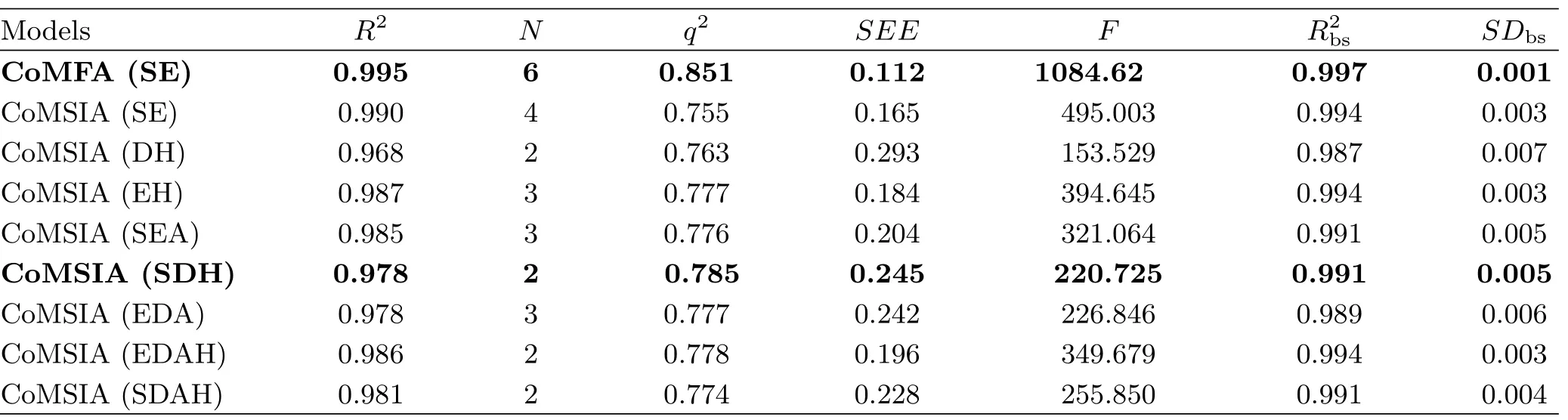
TABLE II Summary of CoMFA and CoMSIA results.
B.3D-QSAR analyses
The 3D-QSAR models of Abl T315I kinase were established from CoMFA and CoMSIA analyses and their statistical results are displayed in Table II.For a reliable predictive model,the cross-validated coefficient q2should be greater than 0.5.
The optimal CoMFA model yielded high R2(0.995), F(1084.620),q2(0.851),as well as small SEE(0.112), indicating a satisfactory internal predictive capability of the model.Moreover,the R2bsof 0.997 and SDbsof 0.001 obtained from bootstrapping analysis for 100 runs further con firm the statistical validity and robustness of the established CoMFA model.The steric and electrostatic contributions were found to be 51.3%and 48.7%, respectively. Therefore,both the steric and electrostatic fields are important factors in fluencing the ligandreceptor interactions and the steric feature contributes more preponderant than electrostatic one to the Abl inhibitory activity.
For the CoMSIA analysis,different combinations of steric(S),electrostatic(E),hydrophobic(H),hydrogen bond donor(D),and acceptor(A) fields were used to seek the best CoMSIA model.As a result,the CoMSIA(SHD)model with the highest q2(0.785),relative higher R2(0.978)and F(220.725)value,and lower SEE(0.245)value were chosen as the best model for further analysis.In addition,the bootstrapping analysis for 100 runs was employed and an R2bsof 0.991 and SDbsof 0.005 were obtained.The contributions of steric,hydrophobic and hydrogen bond donor fields are 0.270,0.473 and 0.257,respectively.At this time,the hydrophobic field makes the largest contribution to Abl inhibitory activity,indicating its crucial role in stabilizing the ligand-Abl receptor complex.
Moreover,an external test set with 14 compounds was employed to evaluate the reliability and reasonability of the built models.The predicted R2predvalues of the CoMFA and CoMSIA(SHD)models for the test set were 0.819 and 0.893,respectively,further demonstrating that both models have the satisfactory predictive ability.The predicted pIC50values and the residual values of compounds for CoMFA and CoMSIA(SHD) are listed in Table II.The comparison of the experimental pIC50values and predicted ones with respect to the CoMFA and CoMSIA(SHD)models is plotted in FIG.3,where most points are evenly distributed around the regression line Y=X,indicating the good quality of two models.
To visualize the results of the best CoMFA and CoMSIA models,the 3D color-coded contour maps were generated and depicted in FIG.4 and FIG.5,using the most potent compound 42 as a reference structure.
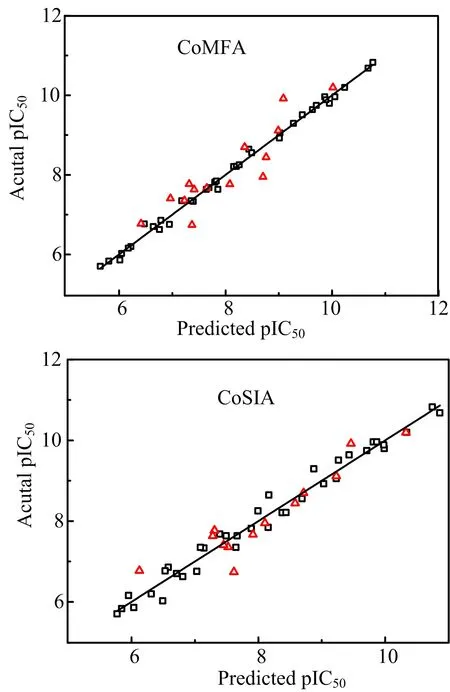
FIG.3 Plots of predicted activities versus actual ones for (a)CoMFA and(b)CoMSIA(SDH)analyses,in which 34 compounds in the training set are expressed as black dots and 14 compounds in the test set are expressed as red triangles.
The CoMFA and CoMSIA(SHD)steric contour maps are shown in FIG.4,where the sterically favorable regions are represented in green and the unfavorable regions in yellow.In FIG.4(a),a green and two yellow contours enclosing the ring-C of substituent R1, which is blocked by the side chains of Val256,Val270 and Ala380,demonstrate that moderate-sized groups on C2′-position and small ones on C3′-and C5′-positions are favorable to the activity.This can be explained by the experimental fact that compounds 9 and 11 with OCH3and OCH2CH3as C2′-substituents,respectively,have higher activities than corresponding compounds 6,7,12,13 with CH3,Cl,OiPr or OPh as C2′-substituent.The higher potency of 28 when compared to 30 is also such a case.Most of the excellent derivatives 27,28,35,39−46 all possess a relative medium-size C2′-substituent,meanwhile,compounds 4, 24 and 25 bearing a C3′-substituent,are the most inactive compounds.Furthermore,there is a small green contour close to the X-position of ring-A,implying that large radius of atom in this position is favorable,which is illustrated by the experimental fact that compounds 27,28,29,33,and 38 with S atom at X-position exhibit over 50 times higher activity than the corresponding compounds 9,11,10,1 and 18 with N atom at X-position.Therefore,we can speculate that it is obvious to improve the bioactivity by possessing S atom in the X-position of ring-A.In addition,it can be easily found that a green and yellow contours lie near the C15 and piperazine ring of substituent R2,demonstrating that a medium-size substituent R2would bene fit the activity.The docking study(FIG.2(a))has shown that the terminal of substituent R2stretches outside the solvent area and thus moderate-sized groups with certain long length could be tolerated by enzyme.That may be the reason that compound 42 possessing longer substituent R2is the most active compound.
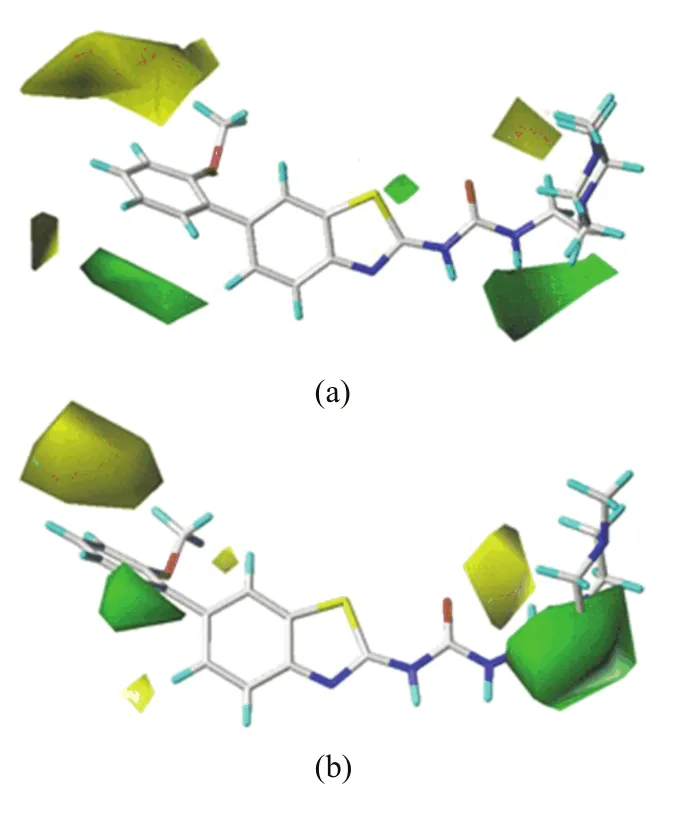
FIG.4 Steric contour maps of most active compound 42 for (a)CoMFA and(b)CoMSIA(SDH)analyses.
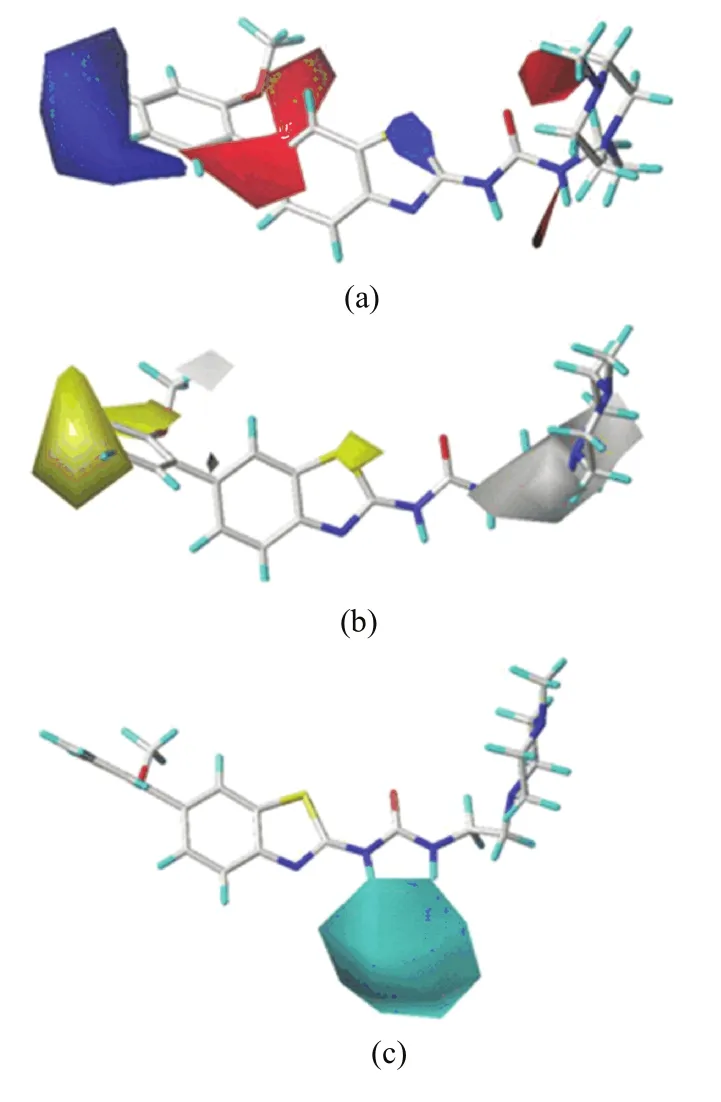
FIG.5 For the most active compound 42:(a)CoMFA electrostatic contour map,(b)CoMSIA(SDH)hydrophobic contour map,(c)CoMSIA(SDH)hydrogen bond donor contour map.
The steric contour map generated by CoMSIA(SHD) (FIG.4(b))is nearly identical to that of CoMFA model apart from a slight difference.There is a small yellow contour near the C5 position of ring-B,implying that a small group at this position would bene fit the bioactivity.The docking result(see FIG.2(a))also shows that the C5-substituent locates on proximity of the side chain of Ala269,suggesting that bulky groups have unfavorable steric hindrance for the receptor.This may explain why compound 31 with-OCH3at this position exhibits almost 10 times worse activity than the C5 unsubstituented compound 27.
On the CoMFA electrostatic field map(FIG.5(a)),the electrostatic interactions are represented by blue and red contours,and the blue contours stand for the electropositive substituents are favorable,while the red contours indicate the electronegative groups are bene ficial.A large blue contour near the C4′-and C5′-positions of ring-C suggests that some electropositive groups at these positions would improve the inhibitory activity.The docking result has shown that the nearest residues are Ala269,Val270,Phe382,which form a hydrophobic pocket.This can be illustrated by the fact that compound 34 with-OCH3at C4′-position displays much lower activity than the corresponding compound 33 with H atom as C4′-substituent.Meanwhile,a small blue contour embedding the X-position of ring-A indicates that introducing a relative high electropositive atom on this position is favorable.Therefore,it can be also used to explain why compounds 27,28,29, 33 and 38 with S atom at X-position exhibit obviously higher activities than compounds 9,11,10,1,and 18 with N atom on this position.There are two large red contours near the first atom of C2′-substituent and C6′-position of ring-C,demonstrating that the electronegative atoms or groups at these positions are favorable. Therefore,compounds 14,16,and 17 have an order for the activity of 14<16<17,with the corresponding C2′-substituent CH3,Cl and OCH3,respectively.Finally, a large and a small red contours are near the N13 and N4′′atoms of ring-D,signifying that the electronegative groups at these sites may enhance the activity.The docking study has shown that the nearest residue toward N13 is Met318,which can form a strong H-bond with N13.In additon,ring-D is partly exposed to the solvent area and thus the R2with N atom should have some good physicochemical properties,such as solubility,permeability,etc.,to increase the binding-ability to the plasma protein.
FIG.5(b)depicts the CoMSIA hydrophobic contour, where yellow contours show hydrophobically favored regions and white contours indicate hydrophilically favored regions.A relatively large yellow contour embedding the C3′-,C4′-and C5′-positions of ring-C,indicates that the hydrophobic groups are well tolerated at these locations.This is consistent with the above result that high electropositive groups on C3′-,C4′-and C5′-substituents are bene ficial to the activity,because usually electropositive groups exhibit relatively hydrophobic. In docking,there is a big hydrophobic pocket enclosing ring-C,implying that these substituents may have many hydrophobic contacts with the receptor.Another small yellow contour covering the X-position of ring-A suggests that the atom with relative hydrophobic character prefers this position.Meanwhile, two white contours were observed above the first atom of C2′-substituent of ring-C and implanting N13 and N1′′of ring-D,suggesting that the hydrophilic atoms in these regions are favored.This may be attributed to electrostatic interactions between the electronegative O atom of C2′-substituent and N1′′atom of ring-D and NH3+group of Asn368 and Asn322,and also the residue Met318 can form a strong H-bond with N13 in docking. This is well in accordance with the above result that higher electronegative groups on these position can enhance the inhibitory activities.
In CoMSIA hydrogen bond donor maps(FIG.5(c)), the cyan contour is favorable to activity,whereas the purple contour is unfavorable to activity.A large cyan contour is found adjacent to the H atoms linking to N10 and N13 atoms,suggesting that hydrogen donor groups at these positions would bene fit the activity.Therefore, H atoms in these positions are likely electron-accepting sites,on which they can form hydrogen bonds with the carbonyl oxygen atom of Met318 in docking.
C.MD simulations
To obtain further information of the ligand-receptor interactions in the binding process,we selected the above two docked complexes(2Z60-42 and 2Z60-25) as the initial conformations to perform the MD simulations.The dynamic picture of the conformational changes of the two complexes is outlined in FIG.6.It can be clearly seen that both systems share a similar root-mean-square displacement(RMSD)behavior.The plots show that both systems reach equilibrium after 1 ns.The mean RMSD values of 2Z60-42 and 2Z60-25 were 1.83 and 1.76˚A,respectively,and the relative RMSD fluctuations were very small.These results suggest that the systems were relatively stable throughout the MD simulations.Moreover,the superimpositions of the average structure of the last 1 ns trajectory and the initial docked structures for both systems is displayed in FIG.7,where the green line represents the initial structure of the docked complex,and the megenta line represents the average MD simulated structure.In FIG.7, we can see the docked structures and the MD average structures were at the same binding pocket,with only slight positional changes,which further veri fied the reasonability and rationality of the docking results.
Furthermore,to further examine the residue contribution of the receptor during the binding process,the analyses of the root-mean-square fluctuation(RMSF) about the residue number of both systems are displayed in FIG.8.From FIG.8,we can clearly note that the fluctuations of residue Gly228 for 2Z60-42 and 2Z6˚0-25 systems were,respectively,up to 6.16 and 6.48A, which means the conformations of two compounds having big vibrations at the region of residue Gly228,due to its locating at the N-terminal of 2Z60.And we can also find that Met318,Tyr253,Val256,Ala269,Val270, Ala380 and Phe382 for both systems were rather steady, because Met318 can form strong hydrogen bonds with two compounds,whereas the latter six residues engaged in the hydrophobic pocket mentioned-above in docking,which can in strong van der Waals contacts with the compound 42 and 25.The active site,includingTyr253,Val256,Val269,Val270,and Phe317-Asn322, has larger conformational drift for 2Z60-25 system than that for 2Z60-42 system.Therefore,we can infer that compound 42 may form stronger interactions with 2Z60 than compound 25,which is compatible with the experimental activities.
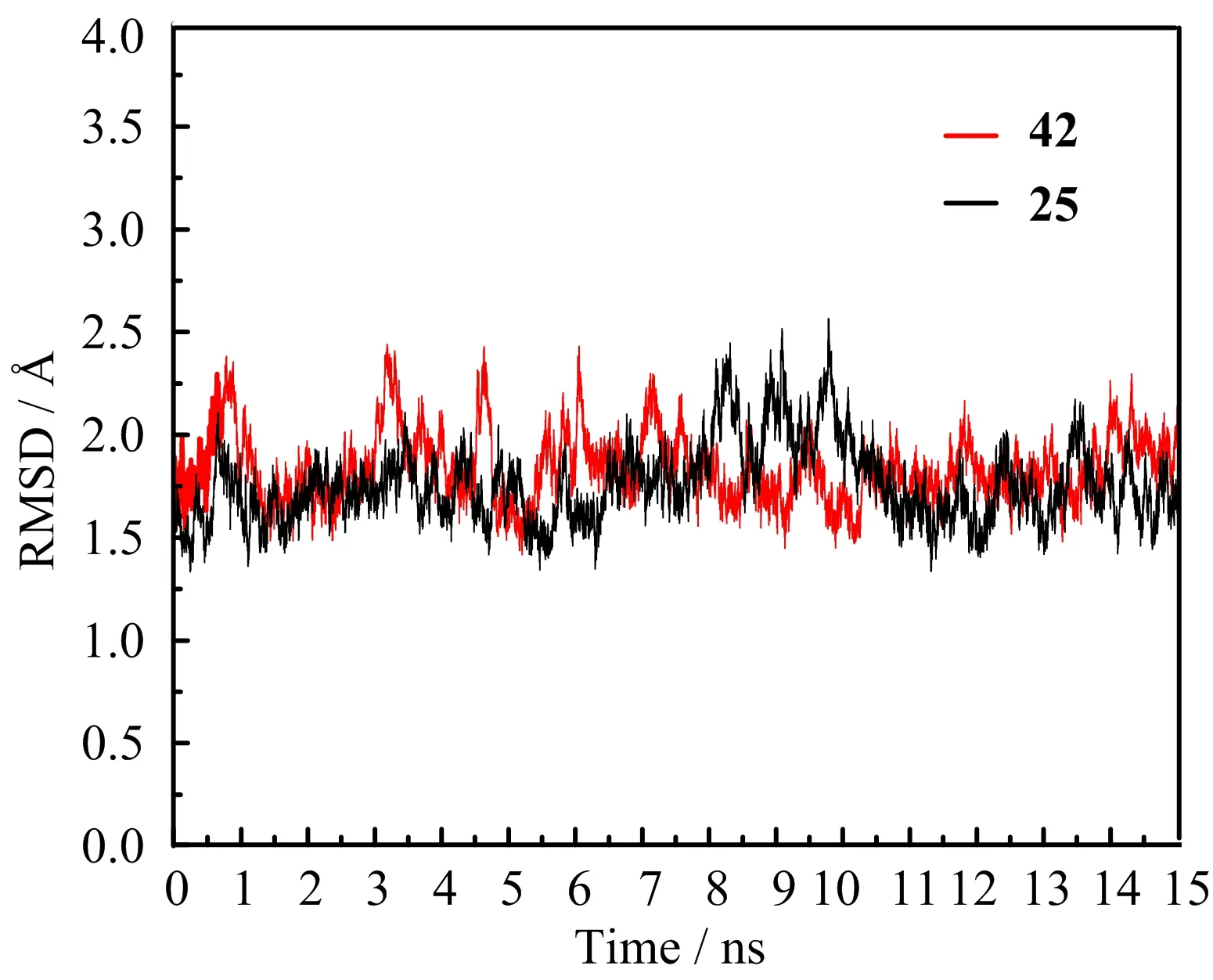
FIG.6 The RMSD values of the backbone atoms of 2Z60-42 and 2Z60-25 systems during the MD simulations.
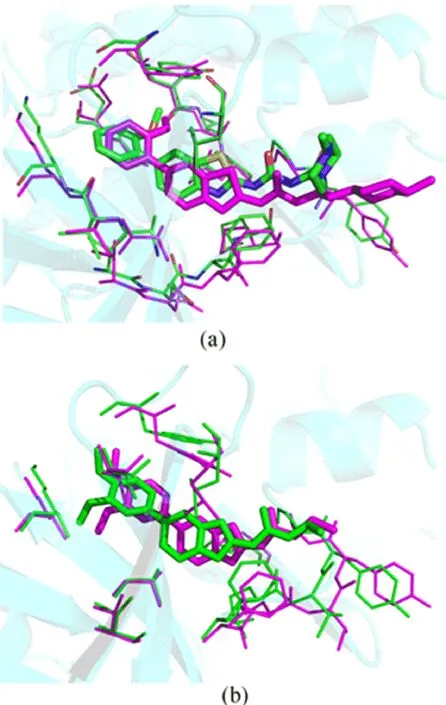
FIG.7 Superimposition of the average structure from the last 1 ns of the MD simulation(magenta)and the initial structure(green).(a)2Z60-42 system,(b)2Z60-25 system.
In addition,the hydrogen bond interaction is also an important force stabilizing the binding conformation. For the two systems,the hydrogen bond interactions from MD simulations are listed in Table III.The hydrogen bond was de fined by distance(<3.5˚A)and orientation(the angle D−H···A>120◦).It was shown that Met318 formed a strong hydrogen bond with compound 42(or 25),with occupancies of over 99%for two systems during the whole MD simulations.But the other two hydrogen bonds existing in initial docking modes have disappeared,due to the slight movement of the conformations of two compounds.
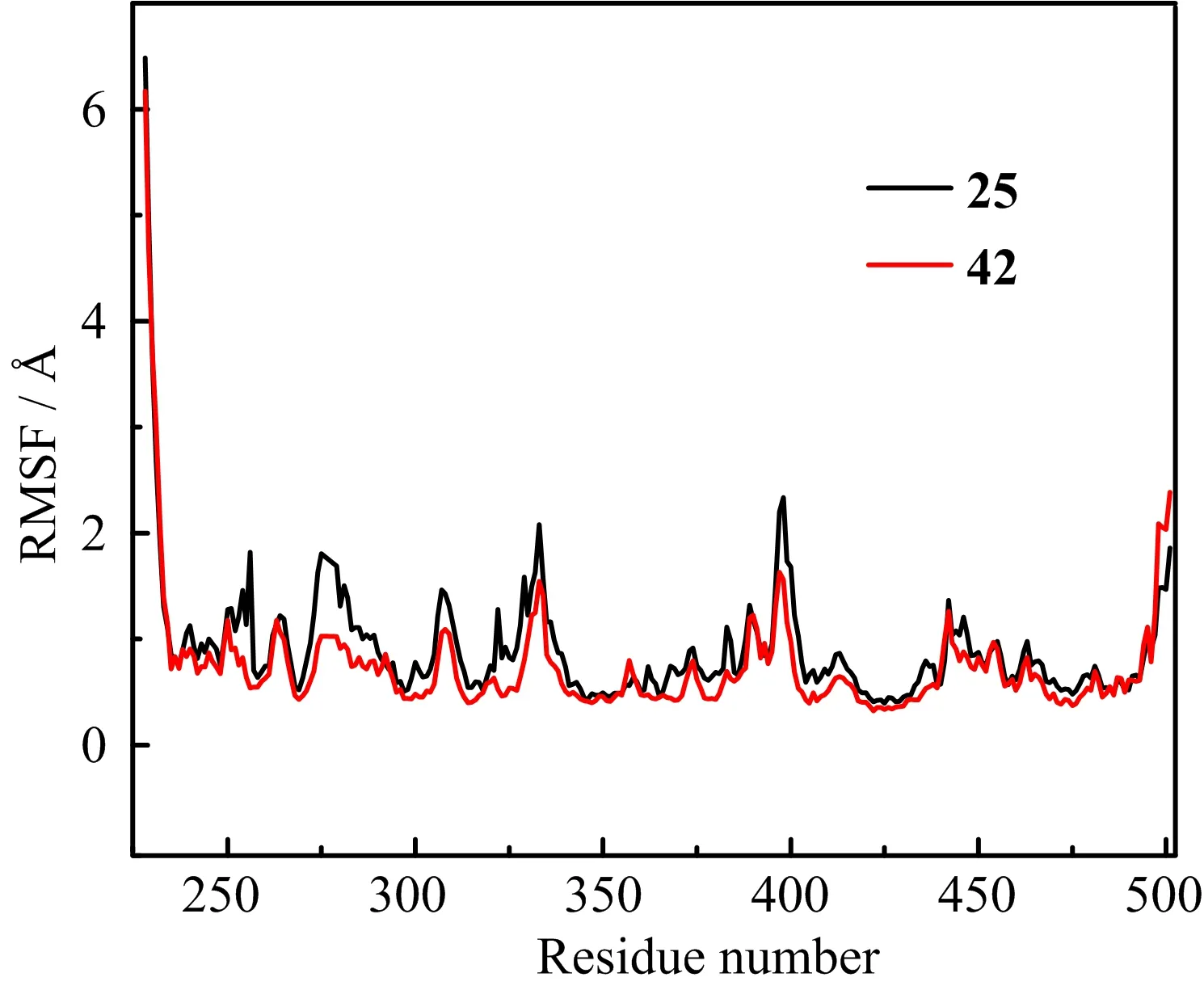
FIG.8 The RMSFs of each residue of the protein for 2Z60-42 and 2Z60-25 systems.

TABLE III Hydrogen bond analysis from the results of MD simulations of 2QOH-42 and 2QOH-25 with donor of ligand@N10 and acceptor of MET318@N-H.
D.Binding free energy analysis
The binding free energies obtained from MM-PBSA calculation for two systems were reported in Table IV. Accordingly,we can observe that the calculated∆Gbindfor the 2Z60-42(−45.07 kcal/mol)was higher than the value of 2Z60-25 system(−42.75 kcal/mol),meaning that the interaction between 2Z60 and compound 42 was stronger than that in 2Z60-25 system,which was in accordance with the order of the experimental bioactivities.According to the energy components of the binding free energies,we speculated that the intermolecular van der Waals interaction(∆Gvdw)and the electrostatic interaction(∆Gele)were the major contributions to the binding.The nonpolar solvation contribution term(∆Gnonpol,sol)contributed slightly favorably for the affinity,whereas polar solvation term(∆Gele,sol)opposed the protein-ligand binding interactions strongly. For both systems,the sum of the van der Waals interaction energy and the electrostatic interaction energy for the 2Z60-42 system(−82.70 kcal/mol)was stronger than that of 2Z60-25 system(−78.60 kcal/mol),which may explain the difference of inhibitory activities for compounds 42 and 25.
In order to reveal the contribution of key residues indetail,the binding free energy in both systems was decomposed to each residue located within 6˚A of the ligand.As shown in FIG.9(a),the residues with the most favorable contributions(higher than 1 kcal/mol)in both systems to the binding free energy are Leu248,Tyr253, Val256,Ala269,Phe317,Met318 and Leu370,and almost all residues energetically contribute more for the binding of compound 42 than that of compound 25.As most of the previous residues are nonpolar,it is reasonable to conjucture that inhibitors can form strong van der Waals interactions with these residues and the van der Waals interactions make major contributions to the binding free energy.Moreover,the electrostatic interactions between Met318 and two compounds which had been validated by docking study and MD simulations, also played key roles in the binding.To represent the results more intuitively,the further energy decomposition of Met318 to two systems was depicted in FIG.9(b).It was revealed that the electrostatic interaction of Met318 contributed slightly favorably to compound 42 than to 25,because compounds 42 and 25 can all form strong hydrogen bond with Met318.In addition,the van der Waals interaction energy of Met318 showed much more favorable contribution to compound 42 than to 25,due to compound 42 more closer to Met318 than compound 25.From these results,we can conclude that hydrophobic interactions play key roles in the binding affinity with compound 42 over compound 25.The formation of a hydrogen bond between inhibitors and the residue Met318 is favarable to stabilize inhibitors in Abl T315I.

TABLE IV Binding free energy(kcal/mol)of both systems.
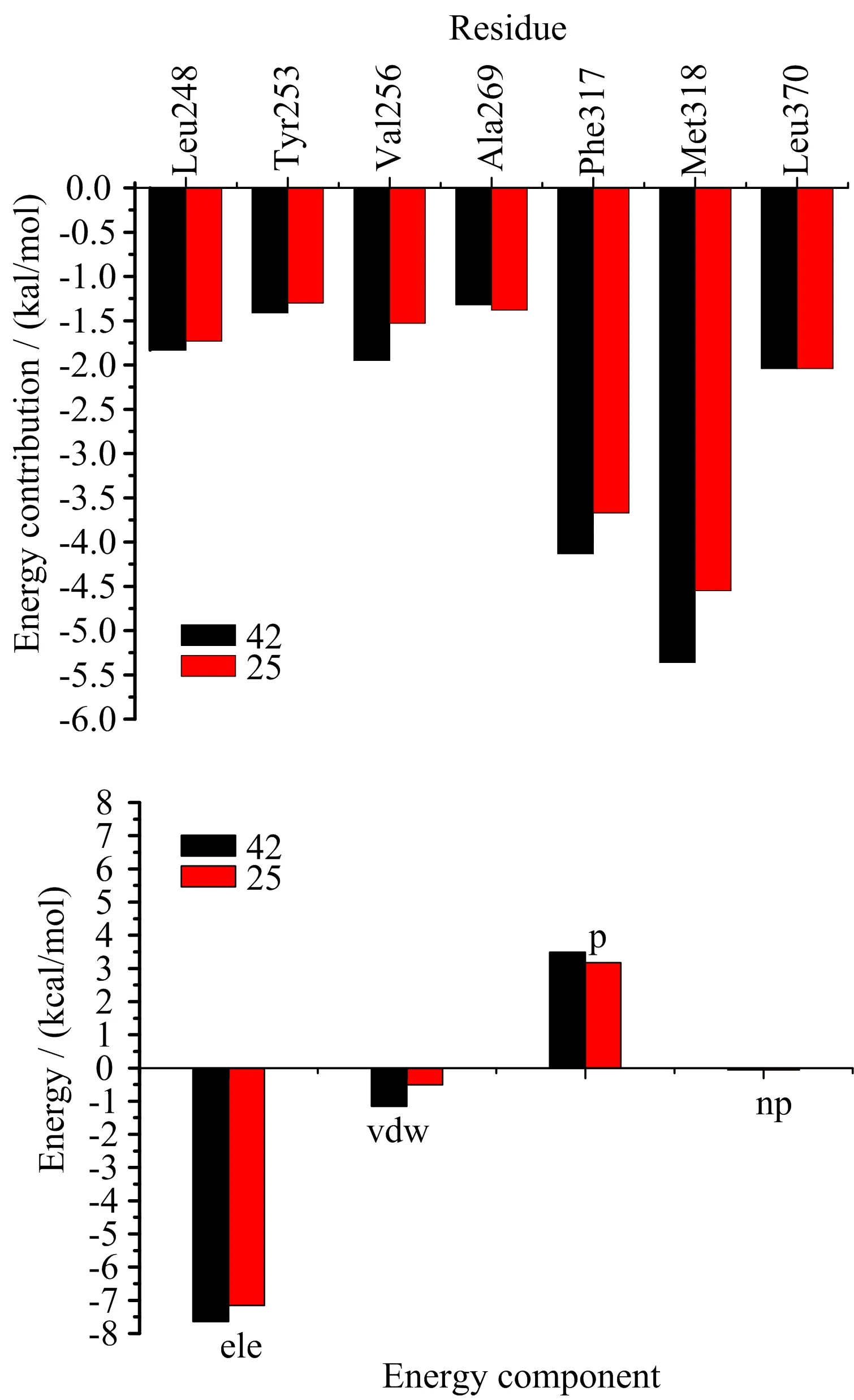
FIG.9 Comparison of energy decomposition for key residues of the two systems.(a)The residues located within 6˚A of the ligand,(b)the residue Met318.ele is electrostatic interaction energy,vdw is van der Waals interaction energy, p is polar solvation free energy,and np is nonpolar solvation free energy.
E.Design of novel compounds with higher inhibitory activity
From the above studies,we found the following points:(i)the compounds with X-position of S atom have higher activity in 48 studied compounds.(ii)The first atoms ofpositions of substituent R1should be electronegative atoms.(iii)Moderate-sized substituent R2with 13-and 1′′-positions being N atoms and its terminal being electronegative and hydrophilic group would enhance the activity.
Based on these findings,we take the most potent compound 42 as a template and start from it to carry out the structural modi fication.The molecular design was focused on the aromatic ring-C and R2substituent, especially on the region beween the 13-and 1′′-positions of R2.We first suggest to adopt-OCH3,NH2or-ONH2instead of H atom linking to the C6′-position of ring-C, because these electronegative groups can reach the red and green areas of 3D-QSAR model.Then,we select NH(CH2)2NHCONH2group as substituent R2,because its N13 and N1′′atoms with strong negative electronic charges just(or almost)fall into the two red and the large white contours.Moreover,we also consider modifying X-position to increase the positive charge with S atom replacing N atom,inspired from the higher activities compounds 27,28,29,33 and 38.Hereby,four new compounds(D1−D4)are designed,their structures and predicted activities(including compound 42 for comparison)by the established 3D-QSAR models are given in Table V.
From Table V,we can find that the predicted activities of compound 42 are very close to its experimental values(pIC50=10.824),and those of the designed compounds(D1−D4)are all much higher (pIC50=10.94−11.88)than that of compound 42 in both the CoMFA and CoMSIA models.The most potential designed molecule D2 exhibits almost 10 times higher activity than compound 42.Such results further suggest that our models have a strong and reliable predictive ability and they can be used in molecular design or structural modi fication.
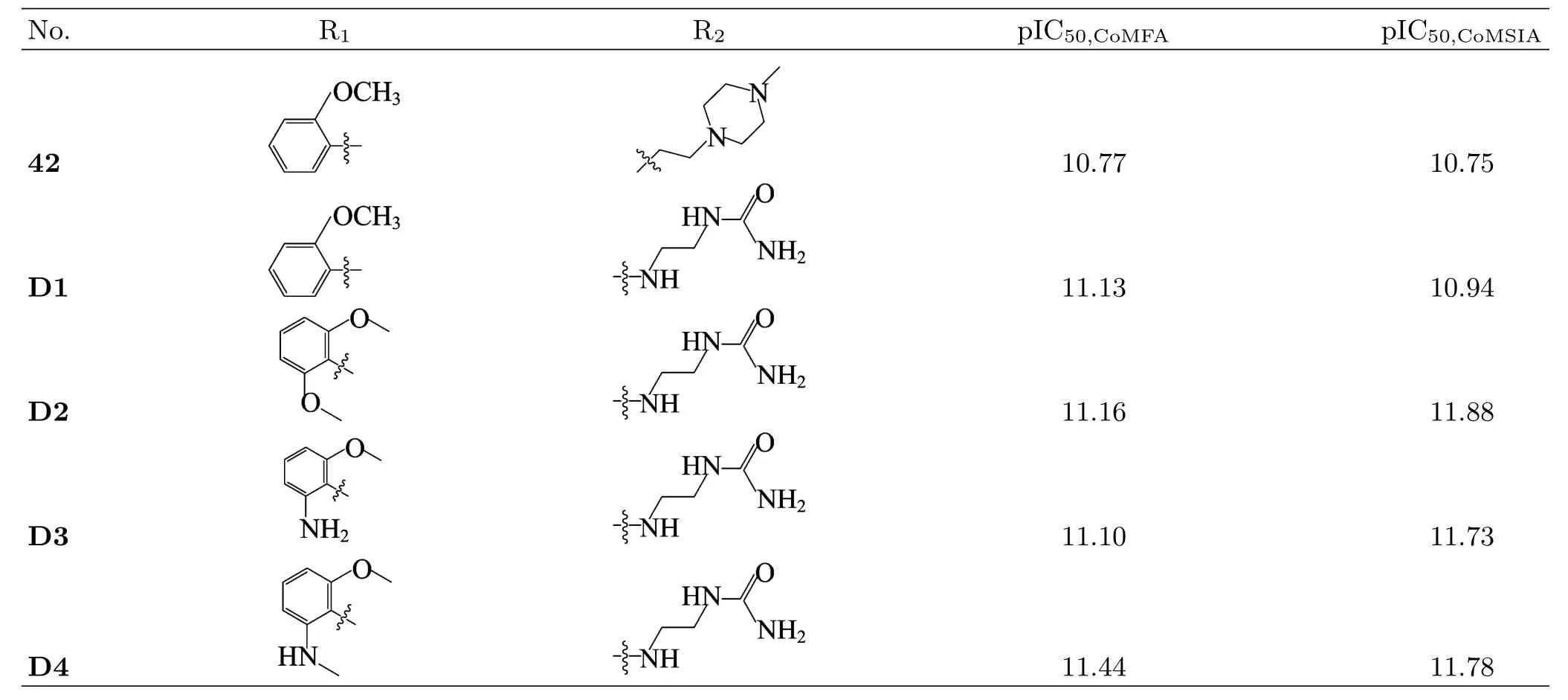
TABLE V Structures and predicted activities of new designed compounds and compound 42.
IV.CONCLUSION
In this work,a multistep computational framework combining molecular docking,3D-QSAR,MD simulations,and MM/GBSA binding free energy calculations was carried out to analyze the interactions between Abl T315I kinase and a series of benzimidazole/benzothiazole based derivatives.The optimal 3DQSAR models exhibited good predictive powers in both the internal and external validations.The docking studies revealed that all compounds bind deeply inside the ATP binding sites with very similar binding modes. The reasonable binding modes of these compounds and the key interaction features were con firmed by the MD simulations.Molecular docking and MD simulations demonstrated that the van der Waals and hydrogen bond interactions are two predominant factors affecting the binding process.The decomposition of binding free energy revealed that the most favorable contributions came from Tyr253,Val256,Ala269,Thr316, Phe317,Met318 and Leu371.Based on the above discussion,four new compounds with quite higher activity have been designed,and their inhibitory activities were also predicted by the established 3D-QSAR models as well as the docking and MD analyses.This work further shows that the multistep computational approaches can provide a detailed understanding of the binding mechanisms between ligand and biotarget,while it can effectively direct the drug-molecular design.
V.ACKNOWLEDGMENTS
This work was supported by the Science and Technology Program of Guangzhou(No.2013J4100071).We also heartily thank the College of Life Sciences,Sun Yat-sen University for the SYBYL 6.9 computation environment support.
[1]S.Faderl,H.M.Kantarjian,and M.Talpaz,Oncology (Williston Park)13,169(1999).
[2]A.Quint´as-Cardama and J.Cortes,Blood 113,1619 (2009).
[3]E.Jabbour,J.Cortes,and H.Kantarjian,Targeted Oncol.4,3(2009).
[4]A.Quint´as-Cardama and J.Cortes,Clin.Cancer Res. 14,4392(2008).
[5]K.Mayer,G.Gielen,W.Willinek,M.M¨uller,and D. Wolf,Leukemia 28,976(2014).
[6]A.Desogus,S.Schenone,C.Brullo,C.Tintori,and F. Musumeci,Expert.Opin.Ther.Pat.4,1(2015).
[7]J.Seymour,D.Kim,E.Rubin,A.Haregewoin,J.Clark, P.Watson,T.Hughes,I.Dufva,J.Jimenez,and F. Mahon,Blood Cancer J.4,e238(2014).
[8]S.Hong,J.Kim,S.M.Yun,H.Lee,Y.Park,S.S. Hong,and S.Hong,J.Med.Chem.56,3531(2013).
[9]G.H.Zeng,W.J.Wu,R.Zhang,J.Xun,W.G.Xie, and Y.Shen,Chin.J.Chem.Phys.25,297(2012).
[10]S.F.Zhou,S.P.Tan,D.Q.Fang,R.Zhang,W.C. Lin,W.J.Wu,RSC Adv.6,85355(2016).
[11]M.Rasheed,C.Richter,L.T.Chisty,J.Kirkpatrick, M.Blackledge,M.R.Webb,and P.C.Driscoll,Biochemistry 53,1092(2014).
[12]Y.F.Ye,C.F.Fu,and S.X.Tian,Chin.J.Chem. Phys.30,25(2017).
[13]S.Version,SYBYL 6.9 CP,St.Louis Tripos Associates, Inc.,(2001).
[14]M.D.M.AbdulHameed,A.Hamza,J.Liu,and C.G. Zhan,J.Chem.Inf.Model.48,1760(2008).
[15]S.J.Cho and A.Tropsha,J.Med.Chem.38,1060 (1995).
[16]R.D.Cramer,D.E.Patterson,and J.D.Bunce,J. Am.Chem.Soc.110,5959(1988).
[17]M.Hao,Y.Li,Y.Wang,Y.Yan,and S.Zhang,J. Chem.Inf.Model.51,2560(2011).
[18]Y.Yang,J.Qin,H.Liu,and X.Yao,J.Chem.Inf. Model.51,680(2011).
[19]D.A.Case,T.Darden,T.E.Cheatham III,C.Simmerling,J.Wang,R.E.Duke,R.Luo,K.M.Merz,D.A. Pearlman,and M.Crowley,AMBER 9,San Francisco: University of California,45(2006).
[20]D.A.Case,T.E.Cheatham,T.Darden,H.Gohlke, R.Luo,K.M.Merz,A.Onufriev,C.Simmerling,B. Wang,and R.J.Woods,J.Comput.Chem.26,1668 (2005).
[21]J.Wang,R.M.Wolf,J.W.Caldwell,P.A.Kollman, and D.A.Case,J.Comput.Chem.25,1157(2004).
[22]A.Jakalian,D.B.Jack,and C.I.Bayly,J.Comput. Chem.23,1623(2002).
[23]U.Essmann,L.Perera,M.L.Berkowitz,T.Darden, H.Lee,and L.G.Pedersen,J.Chem.Phys.103,8577 (1995).
[24]H.C.Andersen,J.Comput.Phys.52,24(1983).
[25]F.Fogolari,A.Brigo,and H.Molinari,Biophys.J.85, 159(2003).
[26]A.Onufriev,D.Bashford,and D.A.Case,Proteins: Structure,Function,and Bioinformatics 55,383(2004).
[27]M.Shen,S.Zhou,Y.Li,P.Pan,L.Zhang,and T.Hou, Mol.Biosyst.9,361(2013).
[28]H.Gohlke,C.Kiel,and D.A.Case,J.Mol.Biol.330, 891(2003).
ceived on April 10,2017;Accepted on May 31,2017)
∗Author to whom correspondence should be addressed.E-mail: wwj@gdpu.edu.cn
杂志排行

CHINESE JOURNAL OF CHEMICAL PHYSICS的其它文章
- γ-Ray Irradiation-Derived MnO/rGO Composites for High Performance Lithium Ion Batteries
- Intermolecular Interactions in Self-Assembly Process of Sodium Dodecyl Sulfate by Vertically Polarized Raman Spectra
- Identi fication of Superoxide O2−during Thermal Decomposition of Molten KNO3-NaNO2-NaNO3Salt by Electron Paramagnetic Resonance and UV-Vis Absorption Spectroscopy
- Highly Responsive and Selective Ethanol Gas Sensor Based on Co3O4-Modi fied SnO2Nano fibers
- Geometric Design of Anode-Supported Micro-Tubular Solid Oxide Fuel Cells by Multiphysics Simulations
- Laser-Assisted Stark Deceleration of Polar Molecules HC2n+1N(n=2,3,4) in High-Field-Seeking State