基于CRISPR/Cas9技术构建猪miR-136基因编辑载体
2017-08-09孙士梅黄世会王嘉福冉雪琴
孙士梅,刘 畅,牛 熙,黄世会,王嘉福*,冉雪琴*
(1.贵州大学 农业生物工程研究院,贵州 贵阳 550025;2.贵州大学 动物科学学院,贵州 贵阳 550025)
·研究报告·
基于CRISPR/Cas9技术构建猪miR-136基因编辑载体
孙士梅1,刘 畅1,牛 熙1,黄世会2,王嘉福1*,冉雪琴2*
(1.贵州大学 农业生物工程研究院,贵州 贵阳 550025;2.贵州大学 动物科学学院,贵州 贵阳 550025)
为了阐明microRNA对猪产仔数的调节机制,采用CRISPR/Cas 9技术构建针对猪miR-136的基因编辑表达载体。通过实验,在miR-136基因成熟区及其下游设计两个单链引导RNA(sgRNA),合成4条磷酸化的单链DNA寡核苷酸,复性后形成两条带粘性末端的双链寡核苷酸片段,插入线性化的载体pX462的BpiI酶切位点处,转化感受态大肠杆菌DH5α,经测序确认,得到两个编辑表达载体pX462-miR-136-1和pX462-miR-136-2。构建了两个编辑miR-136基因的表达载体,为猪繁殖力的调控研究奠定了技术基础。
猪;CRISPR/Cas 9技术;miR-136基因;sgRNA;基因编辑
CRISPR/Cas9技术是一种新型的基因编辑技术。1987年,日本大阪大学研究人员首次发现,大肠杆菌碱性磷酸酶基因的编码区附近含有简单重复序列组成的特殊DNA序列,在单个重复序列之间还插入了间隔序列(space DNA)[1]。随后发现这种间隔成簇短回文重复序列存在于大部分细菌和古细菌中,于2002年被正式命名为“CRISPR/Cas”系统。Cas9是Ⅱ型CRISPR/Cas系统新研发的介导目标DNA切割的核酸酶[2]。2012年,来自霍华德·休斯医学研究所的研究人员发现Cas9蛋白是在crRNA(CRISPR RNA)与tracrRNA(trans-activating chimeric RNA)配对形成的RNA异二聚体的介导下切割目标DNA,他们将这种RNA异二聚体改造为融合成一条sgRNA,以指导Cas9蛋白对靶DNA序列的剪切,从而发展出了CRISPR/Cas9技术[3]。CRISPR是一种来自细菌降解入侵的病毒DNA或外源DNA的免疫机制。在该机制中,Cas蛋白含有两个核酸酶结构域,可以分别切割两条DNA链。一旦与crRNA和tracrRNA结合形成复合物,Cas蛋白发挥核酸酶活性对复合物中的DNA进行切割,造成DNA双链断裂,从而使入侵的外源DNA降解[4]。该技术已经被运用于构建斑马鱼基因敲除细胞系[5]和小鼠动物模型[6]。
MicroRNA是一类非编码的单链小RNA,长度21-25 nt,由70-90 nt并具有发夹结构的单链前体RNA(pre-miRNA)经过Dicer酶的加工形成,通过碱基互补配对与靶mRNA分子的3′-端非编码区(3′-untranslated region,3′-UTR)结合,进而抑制mRNA的后期翻译[7]。MicroRNA在繁殖相关的基因表达、细胞分化等生命活动中起重要的调控作用。
我们在研究猪基因组重测序数据中,从7号染色体132018348-132081671位置发现一个大片段的缺失,经基因注释其中包含miR-136基因。研究发现miR-136的靶基因LHR等对动物的睾丸和卵巢功能起作用[8,9]。为了探索miR-136对猪的繁殖力是否具有调节作用,本文应用CRISPR/Cas9技术构建猪miR-136的基因编辑表达载体。
1 材料与方法
1.1 材料
1.1.1 质粒与菌株 CRISPR/Cas9n质粒pSpCas9n(BB)-2A-Puro (pX462) V2.0(#62987)购自Addgene公司,质粒图谱可于http://www.addgene.org/62987/网站查询。感受态细胞DH5α由贵州大学农业生物工程重点实验室保藏菌株。
1.1.2 试剂 Fast DigestBpiI、FastAP、10×FastDigest Buffer(Green)购自Thermo 公司;Plasmid Safe exonuclease、10×Plasmid Safe Buffer、10 mmol/L ATP购自Epicentr公司:2×T4Ligation Buffer、T4DNA ligase购自Promega公司;AxyPrep DNA凝胶回收试剂盒、质粒小量提取试剂盒购自天根生化科技(北京)有限公司。1.1.3 引物合成与测序 寡链核苷酸(oligo)DNA和引物合成以及测序由上海英潍捷基有限公司完成。1.2 方法
1.2.1 sgRNA oligo序列的设计 根据http://www.mirbase.org/网站确定miR-136基因序列(图1);根据http://crispr.mit.edu/网站设计在miR-136基因序列的成熟区和前体区设计sgRNA序列。sgRNA设计的原则:靶点前面为转录起始信号G,靶点的后面为PAM序列NGG,在其两端加上酶切位点;根据Target Sequence Cloning Protocol标准,在每条sgRNA序列F链的5′端添加CACC,R链的5′端添加AAAC,与pX462质粒经BpiI酶切后形成的粘性末端互补。如F链的5′端第一个碱基不是G,则在5′端CACC下游添加一个G,相应地R链的3′端再增加一个C(表1)。

表1 miR-136 sgRNA oligo和检测引物序列
注:oligo序列上游的酶切位点用小写字母表明,添加的碱基G和C加粗并下划线。

图1 miR-136基因前体序列
Fig.1 miR-136 gene precursor sequence
注:下划线的碱基是miR-136成熟区
1.2.2 重组质粒pX462-miR136-1和pX462-miR136-2的构建 用BpiI对pX462质粒进行酶切,琼脂糖凝胶电泳分离,胶回收线性化的载体。对oligo1、oligo2进行磷酸化修饰和退火。用T4DNA连接酶将线性的pX462载体分别与退火后的oligo1、oligo2连接,反应条件:37℃水浴30 min,16℃连接6-8 h。连接产物转化感受态细胞DH5α,在含氨苄青霉素抗性的LB固体平板上筛选克隆。在pX462载体插入位点的外围设计一对引物UpX462和DpX462(表1),用于筛选阳性菌落。挑取阳性克隆扩大培养,测定插入片段的碱基序列。
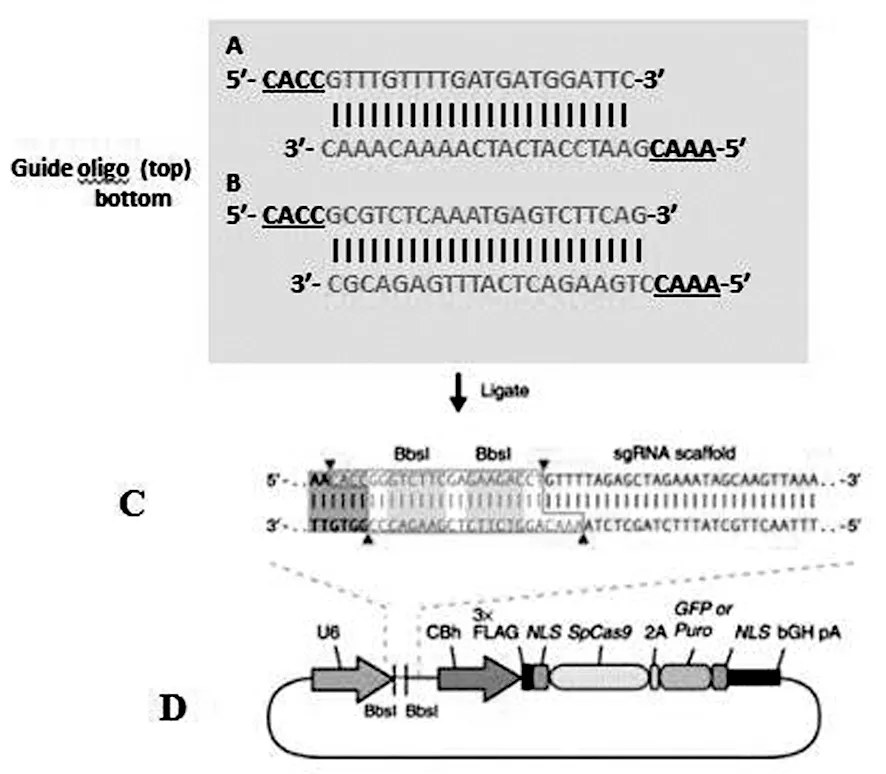
图2 重组质粒pX462-miR136的构建流程
注:A是oligo1磷酸化和退火得到的插入片段;B是oligo2磷酸化和退火得到的插入片段;C是插入片段与线性的pX462载体连接的模式图;D是构建的重组质粒模式图。
2 结果与分析
2.1 oligo1、oligo2的磷酸化和退火
合成oligoF1/R1、oligoF2/R2时分别在单链DNA的5’端增加了磷酸基团修饰。两条单链以1∶1比例混合,在90℃下孵育10 min,室温下自然冷却复性1~2 h。得到双链的寡核酸片段oligo1、oligo2,琼脂糖电泳检测,条带与预期大小相符(图3)。
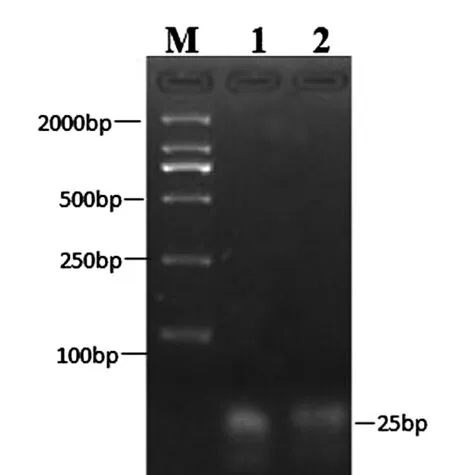
图3 双链的寡核酸片段oligo1、oligo2的检测
注:M为DNA分子量;1泳道是oligo1退火得到的目的片段;2泳道是oligo2退火得到的目的片段。
2.2 pX462质粒酶切
质粒pX462上有BpiI(BbsI)酶切位点(图4),应用BpiI酶切断质粒并去磷酸化,得到线性化的载体。琼脂糖电泳检测约为9 kb,与预期大小一致(图5)。

图4 pX462质粒图谱
注:箭头是BbsI的识别位点。
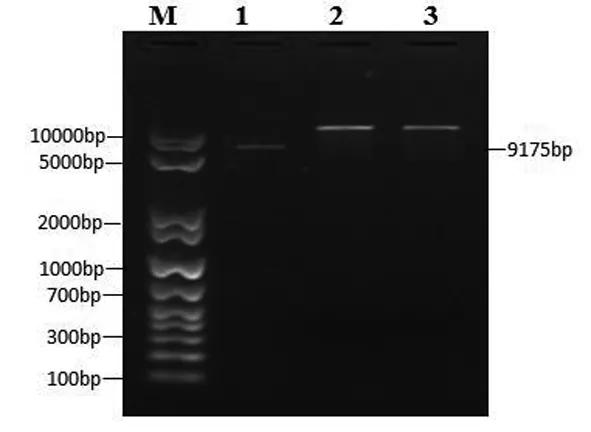
图5 pX462质粒BbsI酶切鉴定
注:M为DNA分子量;1泳道是pX462质粒;2、3泳道是pX462酶切。
2.3 重组质粒pX462-miR136-1和pX462-miR136-2构建
重组质粒pX462-miR136的构建流程如图2所示,按3∶1的比例分别将得到的一对双链的寡核酸片段与线性化载体pX462连接,转化大肠杆菌DH5α,以UpX462/DpX462引物进行PCR检定,得到2#与4#克隆扩增出与预期大小一致的目的片段(图6)。经测序验证(图7、8),在重组质粒的BpiI酶切位点处插入了oligo1和oligo2片段,方向和碱基序列与预期一致,证实oligo1、oligo2正确插入了pX462质粒中,得到重组质粒pX462-miR136-1和pX462-miR136-2。

图6 重组质粒菌落PCR鉴定
注:M为DNA分子量;1泳道是pX462质粒;2泳道是pX462-miR136-1;4泳道是pX462-miR136-2;3、5、6、7泳道阴性克隆。
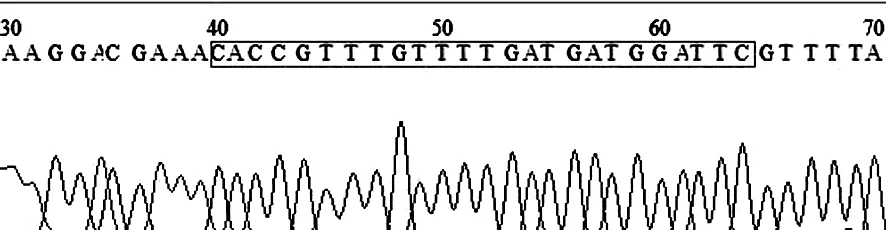
图7 重组质粒pX462 -miR-136-1测序结果
注:方框是插入的正确片段

图8 重组质粒pX462 -miR-136-2测序结果
注:方框是插入的正确片段
3 结论与讨论
本文应用CRISPR/Cas9基因编辑技术构建了miR-136基因编辑的表达载体。应用CRISPR/Cas9基因编辑技术以实现对靶基因的敲除,选择靶基因的识别序列(sgRNA识别序列)是第一步。根据II型CRISPR/Cas9系统的生物学性质,目标基因的识别序列需要是一段20 bp左右的DNA序列,在序列末端紧邻PAM三联核苷酸结构(NGG或者NAG,N代表任何碱基)[10]。本文根据miRNA的作用机理,按照靶点的设计原则,选择miR-136成熟区种子序列与前体序列两个位置[11],设计两条sgRNA,其靶点序列的间距是0~20 bp[12]。
Cas9核酸内切酶有HNH和RuvC两个活性位点,切割后断裂双链DNA,诱发基因组DNA启动同源重组或非同源末端连接机制进行DNA双链的修复,修复过程中可能产生碱基的插入或缺失,改变基因的结构引起突变[13]。CRISPR/Cas9的特异性与跟sgRNA配对且靠近PAM处的7~12 bp有关,因此CRISPR/Cas9的靶向特异性非常低[14]。Ran等将Cas9在RuvC催化位点进行了突变,改造成在sgRNA互补链制造单链切口的切口酶(Cas9n),由一对sgRNA分别引导来实现靶位点的切割,此方法能显著提高基因的敲除效率[15]。
本研究选用改良的pX462质粒,该质粒是为含U6启动子的sgRNA骨架表达载体,表达具有Cas9 D10A切口酶突变的Cas9n(单链切口的核酸酶Cas9)。在酶切pX462质粒时,应用20 uL酶切体系,加入2IU酶(BbsI),10分钟的效果最佳,与插入片段进行连接,转化时效率比较高。成功构建的两个重组表达载体pX462-miR-136-1和pX462-miR-136-2为下一步转染猪繁殖细胞系中奠定了基础。
[1] Ishino Y,Shinagawa H,Makino K,etal.Nucleotide sequence of the iap gene, esponsible for alkaline phosphatase isozyme conversion in Escherichia coli,and identification of the gene product[J].JournalBacteriology,1987,169(12):5429.
[2] Garneau J E,Dupuis M E,Villion M,etal.The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA[J].Nature,2010(468):67.
[3] Jinek M,Chylinski K,Fonfara I,etal.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J].Science,2012,(337):816-21.
[4] MaliP,Esvelt K M,Church G M. Cas9 as a versatile tool or engineering biology[J].NatMethods,2013,10(10):957-963.
[5] Chang N,Sun C,Gao L,etal.Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos[J].CellRes,2013,23(4):465-472.
[6] Wang H,Yang H,Shivalila CS,etal.One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering[J].Cell,2013,153(4):910-918.
[7] Stark A,Brennecke J,Bushati N,etal. Animal MicroRNAs confer robustness to gene expression and have a significant impact on 3’UTR evolution[J].Cell,2005,123(6):1133-1146.
[8] Skye C McIver,Shaun D Roman,Brett Nixon,etal.The rise of testicular germ cell tumours: the search for causes,risk factors and novel therapeutic targets[J].F1000Research,2013(2):55.
[9] Yoshikazu Kitahara,Kazuto Nakamura,Kayoko Kogure,etal.Role of microRNA-136-3p on the Expression of Luteinizing Hormone-Human Chorionic Gonadotropin Receptor mRNA in Rat Ovaries[J].BiologyofReproduction,2013,89(5):114.
[10] Hsu P D,Lander E S,Zhang F. Development and applications of CRISPR-Cas for genome engineering.[J].Cell,2014,157 (6):1262-1278.
[11] Qian Jiang, Xing Meng, Lingwei Meng,etal. Small indels induced by CRISPR/Cas9 in the 50 region of microRNA lead to its depletion and Drosha processing retardance[J].RNABiology,2014,10(11):1243—1249.
[12] F Ann Ran, Patrick D Hsu, Jason Wright1,etal. Genome engineering using the CRISPR-Cas9 system [J].WorldJournalofMedicalGenetics,2014 ,4(3):69.
[13] Jinek M,Chylinski K,Fonfara I,etal.A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity[J].Science,2012,337(6096):816-821.
[14] Pattanayak V,Lin S,Guilinger J P,etal.High- throughput profiling of off- target DNA cleavage reveals RNA- programmed Cas9 nuclease specificity[J].NatBiotechnol,2013,31(9):839-843.
[15] Ran F A,Hsu P D,Lin C Y,etal.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing spec-ificity[J].Cell,2013,154(6):1380-1389.
Construction of Gig miR-136 Gene Editing Vector Based on CRISPR/Cas9 Technique
SUNShi-mei1,LIUChang1,NIUXi1,HUANGShi-hui2,WANGJia-fu1*,RANXue-qin2*
(1.InstituteforAgriculturalBioengineeringofGuizhouUniversity,Guiyang,Guizhou550025,China; 2.FacultyofAnimalScienceandVeterinaryMedicine,GuizhouUniversity,Guiyang,Guizhou550025,China)
The present paper aims to explore the molecular mechanism of microRNA on the litter size of pig. The expressed vector to edit the miR-136 gene was constructed using the clustered regularly interspaced short palindromic repeat (CRISPR) and associated nuclease Cas9 (CRISPR/Cas9) technique. A pair of single guide RNAs (sgRNA) were designed at the miR-136 gene mature sequence and its downstream location based on the precursor of pig miR-136. Four single DNA oligonucleotides were synthesized with a phosphorylation on each strand of the 5`-end, and annealed into two double-strand oligo fragments: oligo 1 and oligo 2. Both of these oligos were inserted into theBpiIsite of plasmid pX462, respectively. After transforming into competent DH5α, the sequences of inserts were confirmed for the two recombinant plasmids as pX462-miR-136-1 and pX462-miR-136-2. Two expression vectors for miR-136 gene editing were constructed, which would be useful for the research of fecundity in pig.
pig; CRISPR/Cas9 technique; miR-136 gene; sgRNA; gene editing
2017-03-08;
2017-05-10
国家高技术研究发展计划(863计划)课题(2013AA102503);贵州省农业攻关项目(黔科合NY[2013]3073号);贵州省“百”层次创新型人才项目(黔科合人才2016-4012号);国家自然科学基金(31672390)。
文献标识码:A
1008-0457(2017)04-0009-04 国际
10.15958/j.cnki.sdnyswxb.2017.04.002
*通讯作者:王嘉福(1962-),男,教授,主要研究方向:生物化学与分子生物学,E-mail:jfwang@ gzu.edu.cn; 冉雪琴(1967-),女,教授,主要研究方向:动物生理与分子生物学;E-mail:xqran@ gzu.edu.cn。
