基于流动注射光度法同时测定催化剂浸渍液中CoO/MoO3
2017-08-09曾明国李永生赵炀杜鑫
曾明国,李永生,赵炀,杜鑫
基于流动注射光度法同时测定催化剂浸渍液中CoO/MoO3
曾明国,李永生,赵炀,杜鑫
(四川大学化学工程学院,四川成都 610065)
基于Mo(Ⅵ)-抗坏血酸、Co(Ⅱ)-KSCN反应体系和流动注射光度法,建立了一个全新的催化剂浸渍液中超高浓度CoO/MoO3的同时测定系统。研究发现:Co(Ⅱ)与KSCN络合物是由K2Co(SCN)4和K4Co(SCN)6构成,利用后者可定量Co(Ⅱ);Co(Ⅱ)对Mo(Ⅵ)-抗坏血酸反应有抑制作用,会干扰Mo(Ⅵ)测定;本研究利用Co(Ⅱ)的抑制作用,人为在测Mo(Ⅵ)的显色剂中加入一定量的硝酸钴,解决了该干扰问题。另外,对测定Co(Ⅱ)/Mo(Ⅵ)用的显色剂中的成分及浓度、进样体积、反应温度等相关影响因素进行了优选,得到的结果是:测定Mo(Ⅵ)的显色剂由15%(质量分数)抗坏血酸、10 g·L-1硝酸钴(以CoO计)及0.1 mol·L-1硫酸组成,测定Co(Ⅱ)的显色剂由37.5% KSCN、0.1 mol·L-1NaAc-HAc (pH 5.8)组成;MoO3和CoO的测定范围分别为10~100 g·L-1和5~50 g·L-1,检出限分别为2.1 g·L-1和1.3 g·L-1, RSD<1.2% (= 11),回收率为98%~104%,分析速度为20样/小时。
钴钼离子;同时测定;流动注射;催化剂;浸渍液;石油;化学分析
引 言
钴钼催化剂有优异的加氢脱硫性能,常用于汽油加氢脱硫精制以提高油品质量[1];同时,其在煤变油以及生物柴油等石油替代品的加氢脱氧精制[2]、Claus反应尾气加氢转化[3]、水煤气变换[4]以及合成氨工业[5]中也有广泛的应用。钴钼催化剂制备常用共浸法,其过程是将g-Al2O3载体浸渍于钼酸铵/硝酸钴混合液内一定时间后,烘干、焙烧而成。由于钼酸铵在水中溶解度较小,但易溶于碱液,因此生产者一般是用一定浓度氨水配制高浓度钼酸铵作为浸渍液,但氨水有腐蚀性且易挥发。研究表明[6-7]:当浸渍液中Co/Mo摩尔比在0.45左右时,就可保证催化剂的催化性能和效率。因此,钼酸铵/硝酸钴共浸液的溶剂可用水溶液替代氨水,这样更利于环保和安全生产。
由于考察催化剂性能时,都用MoO3和CoO来表示浸渍液中钼酸铵和硝酸钴的含量,因此,对于钴钼催化剂生产,MoO3和CoO含量就成了两个重要的监测指标。如果能快速、准确同时测定共浸水溶液中二者的含量,这将对保证催化剂的催化性能及提高催化剂产率具有重要意义。
目前,Co(Ⅱ)、Mo(Ⅵ)的检测方法主要有分光光度法、催化动力学光度法、化学发光法、原子吸收光谱法、原子发射光谱法等。钴/钼的催化动力学光度法主要利用其对某些氧化变色反应的催化作用来间接定量Co(Ⅱ)[8]、Mo(Ⅵ)[9-11]。Co(Ⅱ)的化学发光法[12-14]是利用其对鲁米诺、焦棓酸等发光试剂的氧化发光反应的催化作用来进行定量。但这些方法的测定范围都在微量水平。对于Mo(Ⅵ)化学发光法[15-16],则通常是先将Mo(Ⅵ)还原为Mo(Ⅲ),后者再与鲁米诺在一定条件下产生化学发光。为了精确控制反应,化学发光法通常会与流动注射分析(flow injection analysis, FIA)联用[14-17]。原子吸收、原子发射光谱法[18-25]也常用于痕量钴和钼的测定,并具有精度高、检出限低以及抗干扰能力强等特点,但所需仪器昂贵,不适合工业在线监测。目前测定催化剂中活性组分CoO和MoO3的行标法都是手工光度法[26],其原理是利用Co(Ⅱ)与亚硝基R盐反应生成红色络合物去定量CoO,利用Mo(Ⅵ)还原成Mo(Ⅴ)后再与硫氰酸盐络合生成血红色络合物去定量Mo(Ⅵ);光度法优点是仪器便宜,但操作烦琐,且只能在化学平衡态下测定,分析速度很慢。此外上述诸方法,只能用于微量或痕量Co(Ⅱ)和Mo(Ⅵ)的分析,若要强行用于浸渍液中高浓度Co(Ⅱ)、Mo(Ⅵ)的检测,则要先进行上万倍稀释,这将会导致极大的稀释误差。
因此,基于FIA可定时定量控制反应的特点[27-28],与光度法结合,张红等[29]利用NH4SCN-抗坏血酸-Mo(Ⅵ)反应体系、杜鑫等[30]利用KH2PO4-抗坏血酸-Mo(Ⅵ)反应体系,分别对高浓度Mo(Ⅵ)的FIA测定方法进行了研究,但结果表明,Co(Ⅱ)对Mo(Ⅵ)的测定有干扰,不能用于Co(Ⅱ)/Mo(Ⅵ)共浸液的测定。因此,本文对此进行深入研究,以期解决此问题,并建立一种同时快速自动测定Co(Ⅱ)/Mo(Ⅵ)的方法。
1 实验部分
1.1 仪器及试剂
本实验所用主要仪器有FIA-3100型流动注射处理仪(北京吉天仪器公司)、AUW120D型电子天平(日本岛津公司)、UV-1800PC型紫外可见分光光度计(上海美谱达公司)、DF-101S集热式水浴恒温加热磁力搅拌器(杭州科丰仪器公司)。
本实验所用试剂有(NH4)6Mo7O24·4H2O、98%浓硫酸、冰醋酸、抗坏血酸、Co(NO3)2·6H2O、无水乙酸钠(成都市科龙化工试剂厂)、硫氰酸钾(天津瑞金特化学品公司),均为分析纯。
1.2 试剂配制
测Mo(Ⅵ)用显色剂(R1):称取15.0 g抗坏血酸置于100 ml烧杯中,再加入10.0 ml硝酸钴储备液和5.0 ml硫酸(2 mol·L-1),用水溶解后,转入100 ml容量瓶,定容。
测Co(Ⅱ)用显色剂(R2):称取37.5 g KSCN、0.92 g无水乙酸钠置于100 ml烧杯中用水溶解,再加入0.24 ml冰醋酸,移至100 ml容量瓶内,定容。
钼酸铵储备液(125.0 g·L-1,以MoO3计):称取76.68 g (NH4)6Mo7O24·4H2O于500 ml烧杯中,加适量水溶解,移至500 ml容量瓶中,定容,摇匀。
硝酸钴储备液(100.0 g·L-1,以CoO计):称取194.28 g Co(NO3)2·6H2O置于500 ml的烧杯中,用水溶解,搅拌,完全溶解后移至500 ml容量瓶中,用水定容至刻度。
Mo(Ⅵ)/Co(Ⅱ)混标液:分别取不同体积的上述钼酸铵和硝酸钴储备液,混合配制成6种不同含量混标液,其MoO3和CoO的含量分别为:10 g·L-1+50 g·L-1,30 g·L-1+40 g·L-1,50 g·L-1+30 g·L-1,70 g·L-1+20 g·L-1,90 g·L-1+10 g·L-1,100 g·L-1+5 g·L-1。上述试剂配制用水均为去离子水。
1.3 测定原理
Mo(Ⅵ)测定原理:在酸性条件下,MoO42-缩聚成七聚钼同多酸,然后用抗坏血酸将钼同多酸还原成钼同多蓝,在620 nm处测定其吸光度值定量Mo(Ⅵ)。具体反应方程式如下
7MoO42-+8H++ 4H2O (1)
(钼同多酸)
[Mo(Ⅵ)Mo(Ⅵ)6O24]6-+ C6H8O6
[Mo(Ⅴ)Mo(Ⅵ)6O24]7-+C6H6O6(2)
(钼同多蓝)
Co(Ⅱ)测定原理:在一定条件下,Co(Ⅱ)与KSCN反应生成蓝色络合物,然后在620 nm处测定络合物的吸光度来定量Co(Ⅱ)。
为了确定Co(Ⅱ)/KSCN络合物的络合比,进行了相关实验,得到图1。可以看出,当Co(Ⅱ)浓度不变,逐渐增加KSCN浓度,产物在512、620 nm处有两个吸收峰。以产物在512、620 nm处吸光度值为纵坐标、KSCN浓度为横坐标,得图1内的插图。从该插图可以看出,随着KSCN浓度的增加,512 nm处的吸光度曲线斜率由大逐渐变小,而620 nm处吸光度曲线斜率由小逐渐变大,二者变化趋势不同,说明络合产物中有两种物质。为了进一步确认产物,用斜率比法对512 nm处产物的络合比进行了测定,得到结果是:KSCN过量时Co(Ⅱ)-吸光度的响应曲线方程为= 32.91Co(Ⅱ)+ 0.0052,Co(Ⅱ)过量时去除Co(Ⅱ)背景吸光度值后KSCN-吸光度的响应曲线方程为= 7.55KSCN+ 0.0045,由此得到SCN-/Co(Ⅱ) 络合比为32.91:7.55 ≈ 4:1。用连续变化法和摩尔比率法得到的络合比与此结果一致,所以在512 nm处有最大吸收的物质是K2Co(SCN)4。然后,用斜率比法对620 nm处产物的络合比也进行了测定,结果是:KSCN过量时Co(Ⅱ)-吸光度的拟合曲线方程为= 2.43Co(Ⅱ)+ 0.01,Co(Ⅱ)过量时去除Co(Ⅱ)本身的吸光度后得到的KSCN-吸光度拟合曲线方程为=0.373KSCN+ 0.002,由此得到SCN-/Co(Ⅱ)络合比为2.43:0.37 ≈ 6:1。用连续变化法得到的结果也相同,所以在620 nm处有最大吸收的物质是K4Co(SCN)6。
因此,测定Co(Ⅱ)的反应确定为
Co2++ 6SCN-Co(SCN)64-(620nm) (3)
另外在620 nm处用摩尔比率法测络合比时发现,KSCN继续增加,响应曲线逐渐向上弯曲,甚至当KSCN超过Co30倍以上依然是向上弯曲的趋势,猜想这是式(3)的逆反应所致。随后,用连续变化法的数据计算了该物质的解离度(),得到= 0.122,不稳= 0.019,此数据表明,该络合产物稳定性较差,的确存在逆向解离过程。
1.4 FIA系统及测定过程
同时测定Co(Ⅱ)/Mo(Ⅵ)的FIA系统见图2,测定时按下列程序进行。
P1, P2—peristaltic pump; V—multi-functional value; S—sample; R1, R2— chromogenic agent; SL1, SL2—sampling loop; RL1, RL2—reagent loop; RC—reaction coil; C—current-carrying; Rec.—recorder; L1,L2—connecting tube between quantitative loop and tee joint; W—waste
(1)Mo(Ⅵ)采样 蠕动泵(P1)转动,P2停止,多功能采样阀(Ⅴ)处于“position Ⅰ”;混合样品(S)、测钼用显色剂(R1)在P1的抽吸作用下分别充满样品定量环1(SL1)、Mo(Ⅵ)显色剂定量环1(RL1),多余的混合样品和显色剂排废;与此同时,载流(C)在P1的推动下,经过旁路、反应盘管(RC)进入流通式检测器(D)中,并由其输出一条稳定的空白基线信号,由记录仪(Rec.)记录。
(2)Mo(Ⅵ)注入及Co(Ⅱ)采样 P1停止,P2转动,采样阀转自“position Ⅱ”;载流将“混合样品塞”和“Mo(Ⅵ)的显色剂塞”推入RC中,进行合并及反应,生成与Mo(Ⅵ)相关的有色产物,并流进检测器,在620 nm波长下检测其吸光度值;与此同时,混合样品、测钴用显色剂(R2)又在P2的抽吸作用下充满样品定量环2(SL2)和Co(Ⅱ)显色剂定量环2(RL2)。
(3)Co(Ⅱ)注入及Mo(Ⅵ)采样 P1转动,P2停止,采样阀返回“position Ⅰ”;载流将“混合样品塞”和“Co(Ⅱ)的显色剂塞”推入反应盘管,并进行合并和反应,生成的Co(Ⅱ)/KSCN络合物流入光度检测器,也在620 nm波长下检测;与此同时,混合样品(S)、测钼用显色剂(R1)又第2次在P1的抽吸作用下充满SL1和RL1,为第2次测定做准备。
步骤(2)、(3)循环,实现Co(Ⅱ)、Mo(Ⅵ)的同时测定。
2 实验结果与讨论
本研究设计了一个新的同时测定Co(Ⅱ)、Mo(Ⅵ)的系统,钴/钼检测共用同一个流通式检测器。考虑到合并带法显色剂会被载流稀释,所以先选定了一个较大显色剂定量环体积(RL1和RL2均为220 μl)。然后,在此基础上考察了各参数对灵敏度、响应曲线线性范围以及线性相关系数的影响。由于测定Mo(Ⅵ)时共存的Co(Ⅱ)有负干扰,所以采用饱和法,在测定Mo(Ⅵ)的显色剂中添加了一定量硝酸钴干扰物质,使干扰趋于稳定。
2.1 检测波长确定
图3(a)表明:KSCN在400~800 nm范围内无吸收(曲线1),Co(Ⅱ)在520 nm处有最大吸收(曲线2),二者的络合产物在520、620 nm处都有最大吸收(曲线3、4);为避免Co(Ⅱ)本身颜色的干扰,本系统的测定波长选择在620 nm。另外,钼酸铵溶液本身无色,与Co(Ⅱ)不反应,二者混合后也不影响Co(Ⅱ)络合产物的最大吸收。图3(b)表明:钼酸铵还原产物在600~800 nm范围内有吸收,在730 nm有最大吸收峰。为了能在相同波长下测定Co(Ⅱ)、Mo(Ⅵ),在600~700 nm范围内对Mo(Ⅵ)的线性响应进行了考察,结果显示在此波长范围内Mo(Ⅵ)响应曲线的线性都很好(>0.9990)。综合考虑,最后选定620 nm波长作为Co(Ⅱ)、Mo(Ⅵ)的共用检测波长。
2.2 显色剂成分确定
FIA系统的初始实验条件定为:测Mo(Ⅵ)用的显色剂为10%(质量分数)抗坏血酸 + 0.2 mol·L-1硫酸,测Co(Ⅱ)用的显色剂为25%(质量分数)KSCN + 0.1 mol·L-1NaAc-HAc(pH 5.8);Mo(Ⅵ)注入体积为60 μl,Co(Ⅱ)注入体积为90 μl;系统总流量为2.0 ml·min-1;水浴温度为25℃。
2.2.1 测Mo(Ⅵ)显色剂中Co(Ⅱ)的加入量 80 g·L-1和160 g·L-1(以MoO3计)钼酸铵标液作为测试样品,向其中添加等体积的不同浓度硝酸钴,然后测定对应的吸光度,其结果见图4内插图。可以看出:含Co(Ⅱ)的Mo(Ⅵ)标液比不含Co(Ⅱ)的Mo(Ⅵ)标液响应值低,这表明Co(Ⅱ)的确对Mo(Ⅱ)的测定有负干扰;当Mo(Ⅵ)标液中添加的Co(Ⅱ)达到8.0 g·L-1(10 g·L-1CoO)后,Mo(Ⅵ)-VC产物的吸光度不再受其中Co(Ⅱ)的影响。由此初步选定在测Mo(Ⅵ)显色剂中添加7.9 g·L-1Co(Ⅱ),使Co(Ⅱ)干扰先趋于稳定。
然后,又通过Mo(Ⅵ)单标曲线和Co(Ⅱ)、Mo(Ⅵ)混标曲线的重合度考察了Co(Ⅱ)对Mo(Ⅵ)测定的影响。当两标准曲线重合时,则表明该条件下共存的Co(Ⅱ)不再影响Mo(Ⅵ)的测定。实验结果见图4,黑线表示的是,在测Mo(Ⅵ)显色剂中添加不同的Co(Ⅱ)浓度后得到的Mo(Ⅵ)单标曲线,红线表示的是,相同条件下得到的Co(Ⅱ)/Mo(Ⅵ)混标曲线。图4也表明:测钼的显色剂中Co(Ⅱ)浓度达到7.9 g·L-1(10 g·L-1CoO)后,Mo(Ⅵ)单标曲线与Co(Ⅱ)/Mo(Ⅵ)混标曲线重合。
2.2.2 抗坏血酸浓度的影响 在本研究中,利用抗坏血酸(VC)将钼酸铵还原为蓝色产物。抗坏血酸有弱酸性,可略微调节反应体系的酸度,也可以掩蔽待测样品中的干扰离子Fe3+和Al3+,增强本方法的抗干扰能力。因此,在5%~25%(质量分数)范围内考察了抗坏血酸浓度对Mo(Ⅵ)测定的影响,其他条件不变。实验结果表明:当抗坏血酸浓度较低时,Mo(Ⅵ)响应曲线向下弯曲,而其浓度较高时,响应曲线向上弯曲;Mo(Ⅵ)标曲的线性相关系数()随VC浓度增加呈先增后降的趋势,当VC浓度为15%时>0.999,因此,最后选择测钼显色剂中VC浓度为15%(质量分数)。
2.2.3 Mo(Ⅵ)显色剂酸度 抗坏血酸还原钼酸铵的反应为慢反应,硫酸直接参与反应,其浓度对反应速率影响很大。同时,H+参与了MoO42-聚合生成的反应,优选显色剂溶液酸度,可获得Mo(Ⅵ)测定的最佳灵敏度。因此,在0.05~0.20 mol·L-1浓度范围内,考察了硫酸浓度对MoO3响应曲线的影响。结果表明:当显色剂酸度较低时,响应曲线向下弯曲,而酸度较高时又会向上弯曲;显色剂酸度为0.1 mol·L-1时,>0.999。因此,选定显色剂中硫酸浓度为0.1 mol·L-1。
2.2.4 测Co(Ⅱ)显色剂中KSCN浓度 在25%~40%(质量分数)范围,改变KSCN浓度,考察其对Co(Ⅱ)-KSCN络合物的吸光度及Co(Ⅱ)响应曲线线性的影响。结果表明:Co(Ⅱ)响应曲线的随着KSCN浓度增加而增大,当KSCN浓度达到37.5%以上时,响应曲线线性最好。因此KSCN浓度选取为37.5%(质量分数)。
2.3 Mo(Ⅵ)和Co(Ⅱ)定量环体积优选
FIA系统中进样体积是影响“样品塞”分散度因素之一,从而也影响分析方法的灵敏度。因此,固定其他条件,在45~150 μl范围内,先改变Mo(Ⅵ)样品定量环(SL1)体积,考察了其对Mo(Ⅵ)响应峰高和标曲的影响。结果表明:SL1体积增加,Mo(Ⅵ)响应峰高逐渐增加;当SL1达到90 μl,Mo(Ⅵ)响应峰高增加趋势变缓,同时Mo(Ⅵ)标曲开始向下弯曲,这说明Mo(Ⅵ)进样体积过大导致反应不充分。为了确保较高的灵敏度和标曲的线性,本研究选定90 μl作为SL1的体积。
随后,在60~150 μl范围内优选了Co(Ⅱ)样品定量环(SL2)的体积。结果表明:SL2体积增加,Co(Ⅱ) 的响应峰高是单调增加趋势,且在考察的体积范围内,SL2对Co(Ⅱ)的标曲线性无影响。综合考虑,最终选择90 μl作为Co(Ⅱ)的SL2体积,使Co(Ⅱ)和Mo(Ⅵ)样品定量环体积一致。
2.4 系统流量对测定的影响
FIA系统的流量能够直接影响“样品塞”和“显色剂塞”合并及反应的时间,从而影响反应进行的程度和分析的灵敏度,因此对其进行了考察。
首先固定其他条件,通过调节P1和P2转速来改变系统总流量,在1.46~2.86 ml·min-1范围内考察了流量对Mo(Ⅵ)响应曲线的影响。结果表明:流量较小时则响应曲线向下弯曲,反之向上弯曲;当流量在2.0 ml·min-1时,Mo(Ⅵ)响应曲线线性较好。然后,在1.65~2.72 ml·min-1范围内考察流量对Co(Ⅱ)响应曲线的影响。结果表明:总流量对Co(Ⅱ)响应曲线线性没有影响。在考察的流量范围内,Mo(Ⅵ)、Co(Ⅱ)的响应值均较高,所以,综合考虑,将FIA系统的总流量固定在2.0 ml·min-1(20 r·min-1)。
2.5 反应温度的影响
温度影响反应速率,因此对温度进行了考察。将RC置于恒温水浴中,在8~80℃范围内,先考察了温度对Co(Ⅱ)响应曲线和响应峰高的影响。结果表明:温度升高,Co(Ⅱ)响应峰高有所增加,但增加幅度不大,其响应曲线的线性基本不变化。然后,在7~35℃范围内,考察了温度对Mo(Ⅵ) 响应曲线的影响。结果表明:温度升高,Mo(Ⅵ)的显色产物响应峰增加,同时其响应曲线线性也受到影响;当反应温度低于25℃时,Mo(Ⅵ)响应曲线向下弯曲,温度高于30℃时,Mo(Ⅵ)响应曲线又向上弯曲,反应温度在25℃左右时,Mo(Ⅵ)响应曲线的线性较好。因此,随后实验时,都将本FIA系统的反应温度控制在25℃±1℃。
2.6 干扰实验
在上述优化条件下,考察了工业级钼酸铵、硝酸钴中共存离子以及其他一些常见离子对本方法的干扰。用120 g·L-1MoO3、80 g·L-1CoO两个标液稀释1倍后的吸光度值作为参照,将常见离子溶液与两个标液按1+1方式混合,之后测定对应的吸光度值,并与参照值对比。若添加离子对测定结果的影响小于5%时,可认为该浓度的共存离子不产生干扰。本研究考察了19种离子对Mo(Ⅵ)测定的影响,得到的不产生干扰的最大浓度(g·L-1)分别为:Co2+(100)、Cu2+(0.05)、Al3+(0.5)、Fe3+(0.1)、Na+(4.0)、Ca2+(2.0)、Mg2+(8.0)、Ba2+(0.05)、K+(6.0)、Zn2+(30)、Ni2+(2.0)、Cr3+(0.3)、PO43-(0.02)、Mn2+(10)、Fe2+(0.1)、Cl-(6.0)、NO3-(10)、SO42-(4.0)、SiO32-(0.02)。结果表明:Cu2+浓度过高时,会影响Mo(Ⅵ)的测定,其原因是Cu2+对VC还原钼酸铵具有催化作用[31]。SiO32-和PO43-浓度过高产生干扰的原因是PO43-、SiO32-在酸性条件下会与钼酸铵反应生成磷/硅钼黄,再与抗坏血酸反应生成灵敏度更高的磷/硅钼蓝[32]。除此之外,在工业级钼酸铵中的其他离子都不会产生干扰。
另外,19种离子对Co(Ⅱ)测定的干扰也进行了考察,不产生干扰的最大浓度(g·L-1)分别为:Cu2+(0.064)、Fe3+(4.0)、Ni2+(0.1)、Ba2+(10)、Zn2+(10)、Mn2+(10)、NH4+(80)、Fe2+(0.5)、Al3+(10)、Cr3+(2.0)、Mg2+(10)、Ca2+(10)、Na+(12)、K+(60)、SO42-(10)、NO3-(10)、SiO32-(2.0)、PO43-(10)、MoO42-(150)。结果表明:工业级硝酸钴中共存的相关离子其含量范围都将不干扰Co(Ⅱ)测定,Mo(Ⅵ)也不影响Co(Ⅱ)的测定。
2.7 方法重现性及检出限
经优化后,得到的同时测定Co(Ⅱ)、Mo(Ⅵ)的FIA系统条件为:测Mo(Ⅵ)显色剂组成为15% (质量分数)抗坏血酸 + 10 g·L-1CoO (用硝酸钴配制) + 0.1 mol·L-1硫酸;测Co(Ⅱ)显色剂的组成为37.5%(质量分数)KSCN + 0.1 mol·L-1NaAc-HAc (pH5.8);系统出口总流量为2.0 ml·min-1;反应温度25℃;Co(Ⅱ)、Mo(Ⅵ)注入体积为90 μl;显色剂注入体积为220 μl;反应盘管长度250 cm。该条件下系统的分析速度为20样/小时。
在优化条件下,测定了以CoO及MoO3计算的一系列Co(Ⅱ)、Mo(Ⅵ)混合标液,其结果见图5。可以看出,在5~50 g·L-1CoO浓度范围内,其响应曲线的线性很好(= 0.0089Co+ 0.0603,= 0.9990);在10~100 g·L-1MoO3的浓度范围内,其响应曲线的线性很好(= 0.007Mo– 0.0058,= 0.9992)。
然后,用两个混标液作为测试样品,进行11次重复测定,考察了本系统的重现性。经计算,本系统对于Co(Ⅱ)、Mo(Ⅵ)的相对标准偏差(RSD)都小于1.2%,这表明本系统的稳定性好、测定结果精度也高。经计算,本系统对于Co(Ⅱ)、Mo(Ⅵ)的检出限(3SD/)分别为2.1 g·L-1MoO3和1.3 g·L-1CoO。如果钴钼催化剂共浸液中CoO和MoO3的浓度范围超出本测定范围,可进行预稀释1~2倍后再检测。
2.8 测定模拟浸渍液样品
本测定方法是针对更环保和安全的Co(Ⅱ)/ Mo(Ⅵ)共浸水溶液开发的,所以用水配制了一些模拟钴钼共浸液样品,然后,基于标准曲线法和标准加入修正法(revised standard addition method, RSAM)用本系统测定了这些模拟样品,并进行了加标回收率实验,结果见表1。
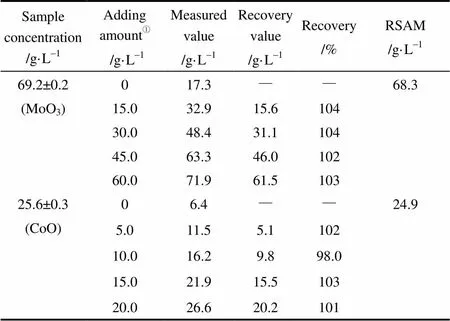
表1 钴钼共浸液样品的回收率实验结果(n=3)
① Sample: standard.
Note: Volume mixing ratio is 1:3.
可以看出,标准曲线法和标准加入法得到的结果一致,回收率在98%~104%,表明本方法测定的浓度值准确可靠,可用于同时测定钴钼催化剂共浸水溶液中的Co(Ⅱ)和Mo(Ⅵ)。
3 结 论
(1)Co(Ⅱ)与KSCN络合物是由K2Co(SCN)4和K4Co(SCN)6构成,K2Co(SCN)4的最大吸收在512 nm,K4Co(SCN)6的最大吸收在620 nm。
(2)Co(Ⅱ)对Mo(Ⅵ)-VC反应有抑制作用。
(3)成功开发了能同时测定共浸水溶液中Co(Ⅱ)/Mo(Ⅵ)的自动分析方法。使用本方法时样品中存在常规含量的Cu2+、Fe3+、Al3+等不产生干扰,钴、钼相互不产生干扰。Co(Ⅱ)和Mo(Ⅵ)的分析范围分别为5~50 g·L-1(以CoO计)和10~100 g·L-1(以MoO3计),分析速度为20样/小时。另外,该系统重现性好、操作简单,可应用于钴钼催化剂的生产过程。
References
[1] FAN Y, SHI G, LIU H Y,. Selectivity enhancement of Co-Mo/Al2O3FCC gasoline hydrodesulfurization catalysts via incorporation of mesoporous Si-SBA-15[J]. Fuel, 2011, 90(5): 1717-1722.
[2] BAO J G, YANG Y Q, WANG W Y,. Preparation and hydrodeoxygenation properties of CoMo/ZrO2-Al2O3catalysts[J].Journal of Fuel Chemistry and Technology, 2011, 39(1): 59-63.
[3] ZHANG K Y, LIU A H, HAO G Y,. Application of CoMo/Al2O3catalyst prepared by wet-mixing kneading method in hydroconversion of Claus reaction tail gas[J]. Petrochemical Technology, 2005, 34(11): 1095-1098.
[4] ZHANG Y F, ZHANG G J, ZHAO Y Q,. Ce-K-promoted Co-Mo/Al2O3catalysts for the water gas shift reaction[J]. International Journal of Hydrogen Energy, 2012, 37(8): 6363-6371.
[5] TSUJI Y, KITANO M, KISHIDA K,. Ammonia synthesis over Co-Mo alloy nanoparticle catalyst preparedsodium naphthalenide-driven reduction[J]. Chemical Communications, 2016, 52: 14369-14372.
[6] PAPADOPOULOU C, VAKROS J, MATRALIS H K,. Preparation, characterization, and catalytic activity of CoMo/g-Al2O3catalysts prepared by equilibrium deposition filtration and conventional impregnation techniques[J]. Journal of Colloid and Interface Science, 2004, 274(1): 159-166.
[7] NIKULSHIN P A, MOZHAEV A V, PIMERZIN A A. CoMo/Al2O3catalysts prepared on the basis of Co2Mo10-heteropolyacid and cobalt citrate: effect of Co/Mo ratio[J]. Fuel, 2012, 100: 24-33.
[8] CHAPARRO L, FERRER L, LEAL L,. A multisyringe flow-based system for kinetic-catalytic determination of cobalt(Ⅱ)[J]. Talanta, 2015, 133: 94-99.
[9] NAKANO S, KAMAGUCHI C, HIRAKAWA N. Flow-injection catalytic spectrophotometic determination of molybdenum (Ⅵ) in plants using bromate oxidative coupling of-hydrazinobensene sulfonic acid with-(1-naphthyl)ethylene diamine[J]. Talanta, 2010, 81(3): 786-791.
[10] MANSOURI A I, MIRZAEI M, AFZALI D,. Catalytic spectrophotometric determination of Mo(Ⅵ) in water samples using 4-amino-3-hydroxy-naphthalene sulfonic acid[J]. Arabian Journal of Chemistry, 2016, 9(S2): 1105-1109.
[11] TEMEL N K, GURKAN R. Catalytic spectrophotometric determination of trace Mo(Ⅵ) in milk-based beverages in the presence of bromophenol blue and H2O2using SDS as a sensitizer[J]. Analytical Methods, 2016, 8(33): 6284-6292.
[12] ZHANG X F, ZHOU Q, LV Y,. Ultrasensitive determination of cobalt in single hair by capillary electrophoresis using chemiluminescence detector[J]. Microchemical Journal, 2010, 95(1): 80-84.
[13] CHEN J Q, YU Y, ZHANG Z W,. NBS-rCDs(OH-) chemiluminescence analysis system for the determination of cobalt ions[J]. Diamond and Related Materials, 2015, 58: 5-9.
[14] SHELLEY R U, ZACHHUBER B, SEDWICK P N,. Determination of total dissolved cobalt in UV-irradiated seawater using flow injection with chemiluminescence detection[J].Limnology and Oceanography-Methods, 2010, 8: 352-362.
[15] DU J X, LI J J, YANG L J,. Sensitive and selective determination of molybdenum by flow injection chemiluminescence method combined with controlled potential eletrolysis technique[J]. Analytica Chimica Acta, 2003, 481: 239-244.
[16] 杨玲娟, 谢天柱, 雷新有. 恒电位电解流动注射化学发光分析法测定钢铁中微量钼[J]. 冶金分析, 2011, 31(11): 24-28. YANG L J, XIE T Z, LEI X Y. Determination of micro molybdenum in steel by constant potential electrolysis - flow injection chemiluminescence analysis[J]. Metallurgical Analysis, 2011, 31(11): 24-28.
[17] GAO X F, IKEBUKURO K, LI Y S,. A novel assay for determination of sulfated bile acids in urine by use of flow-injection chemiluminescence principle with immobilized enzymes[J]. Laboratory Robotics and Automation (Wiley & Sons), 1997, 9(2): 69-79.
[18] ZACHARIADIS G A, THEMELIS D G, KOSSEOGLOU D J,. Flame AAS and UV-VIS determination of cobalt, nickel and palladium using the synergetic effect of 2-benzoylpyridine- 2-pyridylhydrazone and thiocyanate ions[J]. Talanta, 1998, 47(1): 161-167.
[19] SHEGEFTI S, MEHDINIA A, SHEMIRANI F. Preconcentration of cobalt(Ⅱ) using polythionine-coated Fe3O4nanocomposite prior its determination by AAS[J]. Microchimica Acta, 2016, 183(6): 1963-1970.
[20] SHAMSIPUR M, HASHEMI O R, SAFAVI A. Flotation-separation and ICP-AES determination of ultra trace amounts of copper, cadmium, nickel and cobalt using 2-aminocyclopentene-1- dithiocarboxylic acid[J].Analytical Sciences, 2005, 21(9): 1063-1066.
[21] 邵从和. 电感耦合等离子体原子发射光谱法测定铜电积液中锑、铋、钴、镍和砷的含量[J]. 理化检验(化学分册), 2016, 52(6): 691-694. SHAO C H. ICP-AES determination of stibium, bismuth, cobalt,nickel and arsenic in copper electrowinning solution[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis), 2016, 52(6): 691-694.
[22] CANFRANC E, ABARCA A, SIERRA I. Determination of iron and molybdenum in a dietetic preparation by flame AAS after dry ashing[J]. Journal of Pharmaceutical and Biomedical Analysis, 2001, 25: 103-108.
[23] BOSCHETTI W, BORGES A R, DUARTE A T,. Simultaneous determination of Mo and Ni in wine and soil amendments by HR-CS GF AAS[J]. Analytical Methods, 2014, 6(12): 4247-4256.
[24] KAI X M, CHENG L, JIE W,. Determination of W, Mo and other 8 elements in powder metallurgical materials by ICP-AES[J]. Spectroscopy and Spectral Analysis,2007, 27(12): 2578-2580.
[25] 张世龙, 黄启华, 胡小明, 等. 电感耦合等离子体原子发射光谱法测定钨矿石中硅、铁、铝、钛、钨、锡和钼的含量[J]. 理化检验(化学分册), 2016, 52(10): 1237-1240. ZHANG S L, HUANG Q H, HU X M,. ICP-AES determination of Si,Fe,Al,Ti,W,Sn and Mo in tungsten ores[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis), 2016, 52(10): 1237-1240.
[26] 中国工业和信息化部. 有机硫加氢催化剂活性组分分析方法: HG/T 2515—2013[S]. 北京: 化学工业出版社, 2014. Ministry of Industry and Information Technology of the People’s Republic of China. Analytical method of the active composition in organic sulfur hydrogenation catalyst: HG/T 2515—2013[S]. Beijing: Chemical Industry Press, 2014.
[27] LI Y S, GAO X F. Flow Injection Analysis and Application for Chemistry Analysis[M]. Jilin: The Jilin People’s Publisher, 2002: 2-15.
[28] LI Y S, XING C X, YANG L L. Determination of trace-level sodium ion in water-steam system of power plants using an FIA/ISE method with an automatic penetration and alkalization apparatus[J]. Analytical Science,2005, 21(3): 273-279.
[29] 张红, 李永生, 李乔婧, 等. 流动注射-分光光度法测定钴钼催化剂浸渍液中高浓度钼离子[J]. 理化检验(化学分册), 2013, 49(6): 713-719. ZHANG H, LI Y S, LI Q J,. FI-spectrophotometric determination of molybdenum of high concentration in maceration extract of cobalt-molybdenum catalysts[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis), 2013, 49(6): 713-719.
[30] 杜鑫, 李永生. 基于磷钼蓝反应测定钴-钼催化剂浸渍液中钼离子含量[J]. 理化检验(化学分册), 2015, 51(3): 296-299. DU X, LI Y S. Determination of molybdenum ion in impregnation solution of Co-Mo catalysts based on the reaction of phosphorus molybdenum blue[J]. Physical Testing and Chemical Analysis(Part B:Chemical Analysis), 2015, 51(3): 296-299.
[31] 孙宝莲, 李波, 王国栋, 等. 硫氰酸盐光度法测定氨浸渣中的钼[J]. 稀有金属材料与工程, 2009, 38(12): 2253-2255.SUN B L, LI B, WANG G D,. Spectrophotometric determination of molybdenum in the ammonia leaching residue using thiocyanate[J]. Rare Metal Materials and Engineering, 2009, 38(12): 2253-2255.
[32] LI Y S, MUO Y, XIE H M. Simultaneous determination of silicate and phosphate in boiler water at power plants based on series flow cells by using flow injection spectrophotometry[J]. Analytica Chimica Acta, 2002, 455: 315-325.
Simultaneous determination of CoO and MoO3in catalyst impregnation solutions by flow injection spectrophotometry
ZENG Mingguo, LI Yongsheng, ZHAO Yang, DU Xin
(School of Chemical Engineering, Sichuan University, Chengdu 610065, Sichuan, China)
Based on flow-injection analysis (FIA) and reaction systems of Mo(Ⅵ)-ascorbic acid and Co(Ⅱ)-KSCN, a new FIA system was proposed for simultaneous determination of high-concentration CoO/MoO3in impregnation solutions. It was found that reaction products of Co(Ⅱ) and KSCN were K2Co(SCN)4and K4Co(SCN)6, which the latter can be used to quantitate Co(Ⅱ). Co(Ⅱ) inhibitory effect on reaction system of Mo(Ⅵ)-ascorbic acid interfered with the determination of Mo(Ⅵ). Such Co(Ⅱ) interference was resolved by adding cobalt nitrate in the chromogenic agent for Mo(Ⅵ) measurement. In addition, testing conditions were optimized for various factors such as constituents and concentrations in chromogenic reagents for determining Co(Ⅱ) or Mo(Ⅵ), sampling volume and reaction temperature. The chromogenic agents for testing Mo(Ⅵ) were consisted of 15% (mass) ascorbic acid, 10 g·L-1cobalt nitrate (calculated by CoO) and 0.1 mol·L-1sulfuric acid, whereas those for testing Co(Ⅱ) were consisted of 37.5% (mass) KSCN and 0.1 mol·L-1NaAc-HAc (pH 5.8). The detection ranges of MoO3and CoO were in 10—100 g·L-1and 5—50 g·L-1, and the detection limits for MoO3and CoO were 2.1 g·L-1and 1.3 g·L-1, respectively. Relative standard deviation was less than 1.2% (=11), recovery was in range 98%—104%, and the analysis rate was 20 samples per hour.
cobalt and molybdenum; simultaneous determination; flow injection; catalysts; impregnation solution; petroleum; chemical analysis
10.11949/j.issn.0438-1157.20170123
O 657.3
A
0438—1157(2017)08—3056—08
李永生。第一作者:曾明国(1990—),男,硕士研究生。
2017-02-07收到初稿,2017-04-22收到修改稿。
2017-02-07.
LI Yongsheng, lysgxf2005@qq.com
