去除巨噬细胞对血管紧张素Ⅱ诱导的高血压小鼠肾脏的保护作用
2017-07-05刘彦彬林宇涵靳蕊霞周明生
黄 蕾 刘彦彬 林宇涵 靳蕊霞 周明生
去除巨噬细胞对血管紧张素Ⅱ诱导的高血压小鼠肾脏的保护作用
黄 蕾1,2刘彦彬1,2林宇涵1靳蕊霞2周明生1
目的:巨噬细胞浸润与高血压及肾损伤有密切关系,本实验观察使用脂质体氯膦酸二钠(CL)去除巨噬细胞对血管紧张素Ⅱ(AngⅡ)诱导的高血压肾脏损伤的保护作用。 方法:24只雄性C57BL/6小鼠随机分为正常对照、正常+CL组(CL 0.1 ml/10g体重,每周2次尾静脉注射)、Ang Ⅱ组[1.4 mg/(kg·d),通过植入式胶囊渗透压泵连续灌注14d]、AngⅡ+CL组。 结果:与正常组比,AngⅡ明显增加小鼠收缩压,蛋白尿,肾结构损伤和巨噬细胞浸润以及炎症细胞因子肿瘤坏死因子α(TNF-α)、白细胞介素1β(IL-1β)的表达;在AngⅡ高血压小鼠,CL显著降低肾脏巨噬细胞浸润,改善肾脏的结构和功能损伤以及炎症因子的表达,并轻度降低血压。AngⅡ还诱导肾脏纤维化,增加纤维化因子TGF-β1,纤维连接蛋白及NADPH氧化酶gp91phox和p22phox的表达,CL治疗有效地抑制AngⅡ诱导肾脏纤维化以及上述细胞因子的表达。 结论:巨噬细胞浸润在AngⅡ高血压引起的肾损伤中起重要作用,其机制可能与增加巨噬细胞在肾脏组织释放炎症细胞因子及氧化应激反应,去除巨噬细胞对其有保护作用。
肾脏损伤 高血压 血管紧张素Ⅱ 巨噬细胞
高血压是引起肾脏损害和终末期肾病的主要危险因素。 研究表明免疫细胞特别是单核巨噬细胞浸润与高血压肾损伤密切相关。 单核巨噬细胞是机体免疫系统对组织损伤做出反应的第一线免疫细胞,在维持机体内环境功能稳定,调节免疫反应及组织损伤和修复中起着重要的作用。 大量研究表明在各种不同类型肾脏病变中,巨噬细胞是肾组织中浸润最多也是最重要的炎症细胞之一[1]。临床研究也表明在各种肾脏疾病中巨噬细胞在肾脏组织的浸润程度与肾脏组织的、尿蛋白程度、肾功能减退及其预后密切相关[2]。过度的激活肾素血管紧张素系统是引起高血压及高血压肾脏损伤的重要发病机制之一。 血管紧张素 Ⅱ(Ang Ⅱ)是一个前置性炎症调节因子,研究表明 Ang Ⅱ可促进免疫细胞、特别是单核巨噬细胞在肾脏组织中浸润,引起肾脏炎症反应和肾脏组织损伤[1,3-4]。目前还缺乏直接的实验证据证明巨噬细胞在高血压肾损伤中的作用,本研究将利用AngⅡ高血压模型,通过尾静脉注射脂质体氯膦酸二钠(CL)去除巨噬细胞,研究巨噬细胞去除对AngⅡ高血压小鼠肾脏损伤的保护效应,探讨免疫巨噬细胞在高血压及其肾脏损伤中的作用及可能机制。
材料和方法
实验动物 8~10周龄雄性C57BL/6小鼠24只,SPF级,体重20~25g,由北京维通利华实验动物技术有限公司提供,在锦州医科大学实验动物中心所属的SPF级动物房饲养。
主要实验试剂与仪器 AngⅡ(Sigma,RN4474-91-3,A9525-10 mg);植入式胶囊渗透压泵(Durect,CA95014,Alzet model 1007D);CL(美国FormuMax,5 ml);p22phox,gp91phox,Fibronectin(Santa,sc-21870/sc-5827/Sc-9068,100 μl);TGF-β1(R&D,MAB2401);MOMA-2(abcam,ab33451,100 μl);大小鼠无创血压测量系统(北京软隆生物技术有限公司,BP-98A);显微镜(奥林巴斯,BX51);电泳及转印系统(BioRad,PowerPac Basic)。
动物模型构建 雄性C57BL/6小鼠24只常规饲养一周以适应环境。用5%水合氯醛(3 mg/kg I.P.)麻醉,随机分成4组:(1)正常对照组(n=6):在肩胛背部作一假切口并缝合,同时尾静脉注射Liposome(脂质体包裹PBS)作为正常对照;(2)正常+去巨噬细胞组(n=6):在其肩胛部作一假手术,尾静脉注射CL;(3)AngⅡ组(n=6):在肩胛背部植入灌有AngⅡ胶囊渗透压泵,该泵可连续14d释放AngⅡ到周围组织[1.4 mg/(kg·d)],同时尾静脉注射Liposome(脂质体包裹PBS);(4)AngⅡ+去巨噬细胞组(n=6):植入AngⅡ渗透压泵并尾静脉注射CL治疗14d。去巨噬细胞药物CL于手术前一天,以及手术后从尾静脉注射,每周两次,每次注射剂量为0.1ml/10g体重。对照组小鼠在同样时间间隔接受相同剂量的脂质体PBS注射。去巨噬细胞药物CL注射后第2天取尾部血液,制作血涂片进行吉姆萨染色,并以外周血中的粒细胞减少70%作为有效去除巨噬细胞去除的标准[5]。 CL被广泛地应用于去除动物活体内器官和组织中的巨噬细胞,CL经尾静脉进入循环后被巨噬细胞识别、吞噬,进而诱导巨噬细胞调亡[6]。有研究表明通过尾静脉注射CL能有效去除血液循环中75%、组织浸润中70%左右的巨噬细胞[5-7]。
血压、尿蛋白/肌酐比测定 分别测量手术前、手术第7天及手术第14天的收缩压(SBP)。干预结束后收集尿液,测定尿蛋白及肌酐。
肾脏病理分析 新鲜小鼠肾脏组织去除包膜后,沿横截面将肾脏切开,取1/2肾脏组织在4%多聚甲醛固定24h后,包埋、制作石蜡切片。采用PAS、Masson和免疫荧光法染色观察肾脏损伤、胶原沉积及炎症细胞在肾脏的浸润情况。PAS和Masson法的染色程序参照试剂盒说明书(厦门迈威生物科技有限公司、北京雷根生生物技术有限公司)。
肾小球硬化程度评分:每张切片观察20个小球,评分肾小球损伤程度。分为0分:正常肾小球、无硬化;1分:轻度损伤,肾小球损伤比例<25%;2分:中度损伤,肾小球损伤比例25%~75%;4分:完全损伤,肾小球损伤比例>75%。纤维化测量方法:采用IPP软件计算蓝色纤维化面积占总面积的百分比。
用Western Blot的方法分析肾脏组织中炎性细胞因子肿瘤坏死因子α(TNF-α)、白细胞介素1β(IL-1β),纤维连接蛋白(FN)、转化生长因子β1(TGF-β1)及还原型烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶的两个亚基p22phox、gp91phox等的表达。
统计学处理 采用SPSS 17.0软件分析,数据均采用均数±标准差表示,多组间比较采用单因素方差分析(ANOVA),P<0.05为差异有统计学意义。
结 果
SBP、尿蛋白/肌酐 与对照组比较,AngⅡ明显增加动脉SBP(186±7 mmHgvs110±5 mmHg),尿蛋白/肌酐比值(2.6±0.24 mg/mgvs1.8±0.2 mg/mg);巨噬细胞去除药CL治疗明显降低Ang Ⅱ引起的SBP增加(150±6 mmHg,P<0.05,图1A),减少尿蛋白/肌酐比值(2.1±0.16 mg/mg,P<0.05,图1B)。

图1 CL治疗对Ang Ⅱ小鼠血压、尿蛋白/肌酐的影响(n=6)CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素Ⅱ;*:与Ang Ⅱ比较,P<0.05;#:与Ang Ⅱ+CL比较,P<0.05
CL治疗减少了肾脏组织中巨噬细胞的表达 MOMA-2是小鼠巨噬细胞的一个标示性抗原。如图2所示,MOMA-2在肾脏组织中阳性表达,与对照组比较,AngⅡ增加了巨噬细胞在肾脏组织中的浸润;CL治疗降低了巨噬细胞在肾脏组织中的浸润(P<0.05)。
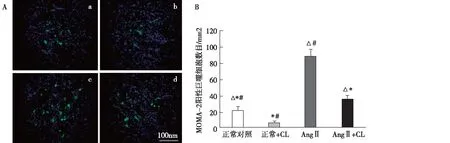
图2 A:MOMA-2在各组小鼠肾脏的荧光表达(IF,×400);B:MOMA-2阳性细胞数目(n=6)Ang Ⅱ:血管紧张素Ⅱ;CL:脂质体氯膦酸二钠;a:正常对照;b:正常+CL;c:Ang Ⅱ;d:Ang Ⅱ+CL;△:与正常+CL比较,P <0.05,*:与AngⅡ比较,P<0.05;#:与AngⅡ+CL比较,P<0.05
CL治疗抑制AngⅡ引起的炎症因子TNF-α、IL-1β的表达 如图3所示,AngⅡ 明显增加了炎症因子TNF-α、IL-1β的表达。CL治疗后高血压小鼠肾脏中TNF-α、IL-1β的表达明显降低。
CL治疗抑制AngⅡ引起的NADPH氧化酶的表达 如图4所示,AngⅡ能显著增加NADPH氧化酶的两个亚基gp91phox和p22phox的蛋白表达量。而去除灌注AngⅡ小鼠巨噬细胞后gp91phox和p22phox的蛋白表达量明显降低,CL治疗能抑制AngⅡ诱导的NADPH氧化酶的表达。

图3 CL治疗对AngⅡ小鼠炎症因子表达的影响(n=6)TNF-α;肿瘤坏死因子α;IL-1β:白细胞介素1β;CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素Ⅱ;*:与Ang Ⅱ比较,P <0.05

图4 CL治疗对AngⅡ小鼠NADPH氧化亚基表达的影响(n=6)CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素Ⅱ;*:与AngⅡ比较,P<0.05;#:与AngⅡ+CL比较,P<0.05
CL治疗抑制AngⅡ引起的肾脏纤维化 TGF-β1及其下游分子FN是调节细胞基质和纤维化反应过程的重要纤维因子。AngⅡ明显增加了FN和TGF-β1的表达,而CL治疗能明显抑制高血压小鼠肾脏中FN和TGF-β1的表达(图5)。
肾脏病理结果
PAS染色 实验2周后,PAS 染色显示泵入AngⅡ的小鼠肾脏出现明显肾小球膨大,肾小管扩张,肾间质增多。AngⅡ组小鼠经CL治疗后,肾脏病理明显改善(图6)。
Masson染色 如图7所示,与其余各组比较,AngⅡ组小鼠肾小球硬化伴肾间质纤维化明显。AngⅡ组小鼠经CL治疗后,纤维化面积明显减少,差别有统计学意义(P<0.05)。

图5 CL治疗对AngⅡ小鼠纤维化的影响(n=6)FN:纤维连接蛋白;TGF-β1:转化生长因子β1;CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素 Ⅱ;*:与AngⅡ比较,P<0.05;#:与AngⅡ+CL比较,P<0.05
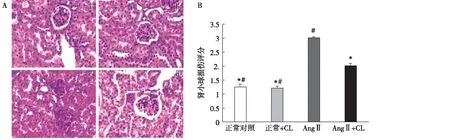
图6 A:各组小鼠肾脏病理变化(PAS,×400);B:肾小球损伤评分(n=6)CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素Ⅱ;a:正常对照;b:正常+CL;c:Ang Ⅱ;d:Ang Ⅱ+CL;*:与Ang Ⅱ比较,P<0.05;#:与Ang Ⅱ+CL比较,P<0.05
讨 论
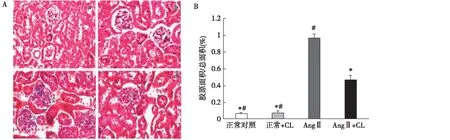
图7 A:各组小鼠肾脏病理变化(Masson,×400);B:纤维化面积百分比(n=6)CL:脂质体氯膦酸二钠;Ang Ⅱ:血管紧张素 Ⅱ;a:正常对照;b:正常+CL;c:Ang Ⅱ;d:Ang Ⅱ+CL;*:与Ang Ⅱ比较,P<0.05;#:与Ang Ⅱ+CL比较,P<0.05
巨噬细胞浸润与高血压肾损伤有密切关系,但目前尚无直接的实验证据证明巨噬细胞在高血压肾损伤中的确切作用。本实验通过尾静脉注射CL对AngⅡ高血压鼠进行巨噬细胞去除,结果表明CL能有效地减少循环血中的单核细胞和肾脏中的巨噬细胞浸润,巨噬细胞去除明显地减轻AngⅡ引起的肾脏结构和功能损伤,同时降低AngⅡ引起的肾脏炎性细胞因子表达和NADPH氧化酶活性。这些结果提供了直接的实验证据支持巨噬细胞参与AngⅡ高血压肾脏损伤的病理机制。
AngⅡ诱导的高血压常伴有肾巨噬细胞浸润增加,浸润的巨噬细胞在肾组织中释放炎性细胞因子引起肾脏结构和功能的损害。TNF-α和IL-1β是巨噬细胞释放的主要炎性细胞因子,也是炎性巨噬细胞被激活(M1型)的标志[8]。目前有研究表明巨噬细胞来源的TNF-α在高血压和糖尿病肾损害中起着非常重要的作用[9-10]。本实验结果显示AngⅡ在增加巨噬细胞在肾组织浸润的同时,肾组织中的炎性细胞因子TNF-α和IL-1β等的表达明显增加,巨噬细胞去除在降低AngⅡ引起的肾结构和功能损伤的同时,也降低了肾组织中这些细胞因子的表达。这些结果提示AngⅡ增加肾组织炎性因子表达和炎症反应至少部分与增加肾组织巨噬细胞浸润有关,AngⅡ通过增加巨噬细胞来源的炎性因子促进肾损伤。
AngⅡ高血压肾病也与氧化应激损伤密切相关[11]。AngⅡ可直接激活肾组织细胞中的NADPH氧化酶,或通过增加浸润在肾脏组织中的炎症细胞(主要是M1型巨噬细胞)NADPH氧化酶诱导肾氧化应激反应[1,12-13]。膜亚基gp91phox和p22phox是NADPH氧化酶在吞噬细胞的主要表达类型[14]。我们的实验结果显示AngⅡ明显增加肾脏组织中NADPH氧化酶的两个亚基gp91phox和p22phox的表达;去除巨噬细胞可降低肾gp91phox和p22phox的表达,表明巨噬细胞是AngⅡ促进肾氧化应激反应的主要来源。
AngⅡ高血压肾损伤的另一个特征是肾脏纤维化, AngⅡ促进肾纤维化主要是通过上调肾TGF-β1[15],TGF-β1是一个重要的纤维调节因子,它主要通过增加基质蛋白的合成来刺激内皮细胞在肾小球系膜积聚[16],在高血压肾脏损害中扮演重要角色[17-18]。肾组织中的TGF-β1可来自肾细胞本身,也可来自浸润的巨噬细胞[1]。我们的实验结果显示巨噬细胞去除明显的改善AngⅡ高血压小鼠的肾纤维化并降低肾组织TGF-β1和FN的表达,这些结果说明巨噬细胞也参与AngⅡ引起的高血压肾纤维化过程。
综上所述,本研究表明巨噬细胞参与了AngⅡ高血压肾脏损伤和肾纤维化病理过程。巨噬细胞去除减少肾脏组织中的巨噬细胞募集及巨噬细胞来源的炎性细胞因子,降低肾脏的炎症,氧化应激反应以及肾纤维化过程,从而降低高血压肾脏组织损伤。 我们的研究结果有可能为发展一个以抑制巨噬细胞肾浸润或巨噬细胞分泌炎性因子为靶目标的高血压肾病治疗方法提供新思路。
1 陈丹,吴义超.巨噬细胞与肾脏疾病.肾脏病与透析肾移植杂志,2004,13(3):256-260.
2 Silva GE,Costa RS,Ravinal RC,et al.Renal macrophage infiltration is associated with a poor outcome in IgA nephropathy.Clinics (Sao Paulo),2012,67(7):697-703.
3 Li JJ,Fang CH,Hui RT.Is hypertension an inflammatory disease? Med Hypotheses,2005,64(2):236-240.
4 Mehta PK,Griendling KK.Angiotensin II cell signaling:physiological and pathological effects in the cardiovascular system.Am J Physiol Cell Physiol,2007,292(1):C82-97.
5 Falkenham A,de Antueno R,Rosin N,et al.Nonclassical resident macrophages are important determinants in the development of myocardial fibrosis.Am J Pathol,2015,185(4):927-942.
6 Van Rooijen N,Sanders A.Liposome mediated depletion of macrophages:mechanism of action,preparation of liposomes and applications.J Immunol Methods,1994,174(1-2):83-93.
7 Zandbergen HR,Sharma UC,Gupta S,et al.Macrophage depletion in hypertensive rats accelerates development of cardiomyopathy.J Cardiovasc Pharmacol Ther,2009,14(1):68-75.
8 Wang Y,Li Y,Wu Y,et al.5TNF-α and IL-1β neutralization ameliorates angiotensin II-induced cardiac damage in male mice.Endocrinology,2014,155(7):2677-2687.
9 Sun L,Kanwar YS.Relevance of TNF-α in the context of other inflammatory cytokines in the progression of diabetic nephropathy.Kidney Int,2015,88(4):662-665.
10 Awad AS,You H,Gao T,et al.Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury.Kidney Int,2015,88(4):722-733.
11 Evans RG,Head GA,Eppel GA,et al.Angiotensin II and neurohumoral control of the renal medullary circulation.Clin Exp Pharmacol Physiol,2010,37(2):e58-69.
12 Ruiz-Ortega M,Esteban V,Rupérez M,et al.Renal and vascular hypertension-induced inflammation:role of angiotensin II.Curr Opin Nephrol Hypertens,2006,15(2):159-166.
13 Harrison DG,Guzik TJ,Lob HE,et al.Inflammation,immunity,and hypertension.Hypertension,2011,57(2):132-140.
14 Forman HJ,Torres M.Reactive oxygen species and cell signaling:respiratory burst in macrophage signaling.Am J Respir Crit Care Med,2002,166(12 Pt 2):S4-8.
15 Xu J,Carretero OA,Liao TD,et al.Local angiotensin II aggravates cardiac remodeling in hypertension.Am J Physiol Heart Circ Physiol,2010,299(5):H1328-1338.
16 Zhou MS,Schuman IH,Jaimes EA,et al.Renoprotection by statins is linked to a decrease in renal oxidative stress,TGF-beta,and fibronectin with concomitant increase in nitric oxide bioavailability.Am J Physiol Renal Physiol,2008,295(1):F53-59.
17 Dahly AJ,Hoagland KM,Flasch AK,et al.Antihypertensive effects of chronic anti-TGF-beta antibody therapy in Dahl S rats.Am J Physiol Regul Integr Comp Physiol,2002,283(3):R757-767.
18 Matsuda H,Fukuda N,Ueno T,et al.Development of gene silencing pyrrole-imidazole polyamide targeting the TGF-beta1 promoter for treatment of progressive renal diseases,J Am Soc Nephrol,2006,17(2):422-432.
(本文编辑 青 松)
Renal protection of macrophage depletion in angiotensin Ⅱ induced hypertensive mice
HUANGLei1,2,LIUYanbin1,2,LINYuhan1,JINRuixia2,ZHOUMingsheng1
1DepartmentofPhysiology,JinzhouMedicalUniversity,Jinzhou121000,China2TheFirstAffiliatedHospital,JinzhouMedicalUniversity;Jinzhou121000,China
Correspondingauthor:ZHOUMingsheng(E-mail:zhoums1963@163.com)
Objective:Macrophage infiltration is tightly contacting with hypertensive renal injury, the present study investigated renal protection of macrophage depletion by clodronate liposome (CL) in angiotensin (Ang) Ⅱ-induced hypertension mice. Methodology:Twenty-four C57BL/6 mice were randomly divided into four groups: normal control, normal plus CL treatment (CL, 0.1 ml/10g BW, 2 times/week by tail vein injection), AngⅡ infusion [1.4 mg/(kg·day), implanted by mini-pump], and AngⅡ plus CL treatment for 14 days. Results:Compared with control mice, AngⅡ infusion significantly increased systolic blood pressure (SBP), proteinuria, renal structural damage, renal macrophage infiltration associated with higher expression of proinflamamtory cytokine tumor necrosis factor (TNF) α and inteleukin (IL) 1β. CL markedly reduced renal macrophage infiltration, renal structural and functional impairment, and the expression of the proinflammatory cytokines with a mild reduction in SBP in AngⅡ mice. Furthermore, AngⅡ also induced renal fibrosis with increasing expression of fibrotic factors TGF-β1 and fibronectin as well as the expression of NADPH oxidae subunits gp91phox and p22phox, depletion of macrophage by CL effectively reversed renal fibrosis and the changes in those molecules induced by AngⅡ. Conclusion: Macrophage was the main contributor to AngⅡ hypertensive renal injury. The underlying mechanisms may involve increased production/release of macrophage-derived inflammatory cytokine and oxidative stress, macrophage depletion protects AngⅡ induced renal hypertensive injury.
renal damage hypertension angiotensinⅡ macrophage
10.3969/cndt.j.issn.1006-298X.2017.03.008
国家自然科学基金项目(81670384,81470532)
1锦州医科大学生理教研室(锦州,121000);2锦州医科大学附属第一医院
周明生(E-mail:zhoums1963@163.com)
2016-09-27
ⓒ 2017年版权归《肾脏病与透析肾移植杂志》编辑部所有
