浙江省三地区冷鲜鸡中大肠埃希菌耐药谱测定及MLST分子分型分析
2017-07-03何祥祥肖英平钱鸣蓉张巧艳
杨 华,何祥祥,3,肖英平,钱鸣蓉,张巧艳,唐 标,*
(1.浙江省农业科学院 农产品质量标准研究所,浙江 杭州 310021; 2.浙江省植物有害生物防治省部共建国家重点实验室培育基地,浙江 杭州310021; 3.西北农林科技大学 食品科学与工程学院,陕西 杨凌 712100)
浙江省三地区冷鲜鸡中大肠埃希菌耐药谱测定及MLST分子分型分析
杨 华1,2,何祥祥1,2,3,肖英平1,2,钱鸣蓉1,2,张巧艳1,唐 标1,2,*
(1.浙江省农业科学院 农产品质量标准研究所,浙江 杭州 310021; 2.浙江省植物有害生物防治省部共建国家重点实验室培育基地,浙江 杭州310021; 3.西北农林科技大学 食品科学与工程学院,陕西 杨凌 712100)
大肠埃希菌是食品中普遍污染的条件致病菌,其耐药性非常严重,耐药基因具有极大的传播风险。为了解浙江省地区禽源大肠埃希菌耐药性特点及分布规律,试验从浙江省3个地区市售冷鲜鸡中分离了59株大肠埃希菌并测定了耐药谱,发现有24种耐药谱,显示了分离株耐药类型的多样性。同时对分离株进行了Multilocus sequence typing (MLST)分子分型,获得38个已知ST型,并发现2个新的ST型。在此基础上,针对7个看家基因进行了系统发育分析,分离株显示了一定的地区差异性。该试验结果可为浙江省禽源大肠埃希菌的耐药性评价和溯源分析提供参考依据。
大肠埃希菌;细菌耐药性;多位点序列分型(MLST);系统发育树
细菌耐药性被认为是人类和动物健康的重要威胁[1-2]。耐药基因可以通过可移动元件(质粒、转座子等)在不同的病原菌中转移[3-7]。多重耐药的革兰阴性菌的耐药性状况尤其严峻,是重要的耐药基因库[8-11]。大肠埃希菌是肠杆菌科中典型的污染指标菌,其耐药表型多样,多重耐药日益严重[12]。禽产业链污染大肠埃希菌溯源分析对于明确致病性大肠埃希菌的污染状况和来源具有重要意义[10],而以方便快速、分辨率高为特征的多位点序列分型(multilocus sequence typing, MLST)技术已经成为微生物的分子流行病学和溯源研究的重要手段[13-14]。
本试验选择对浙江省3个地区市售的冷鲜鸡肉样本进行大肠埃希菌分离,检测耐药谱并对分离株进行MLST分型分析。以进一步探明浙江省地区禽源大肠埃希菌耐药性特点及分布规律,为浙江省禽产业链大肠埃希菌污染防控提供依据。
1 材料与方法
1.1 材料
1.1.1 样品采集
冷鲜鸡整只鸡肉样品来源于浙江省绍兴(SX)、湖州(HZ)以及金华(JH)三个地区的超市或农贸市场,分别采集了10、9和16个,采样时间为2016年8月。大肠埃希菌质控菌株ATCC 25922为实验室保藏,来源于美国典型培养物保藏中心(American Type Culture Collection, ATCC)。
1.1.2 主要仪器与试剂
细菌鉴定及耐药谱分析采用梅里埃VITEK 2 Compact 全自动细菌鉴定及药敏分析系统;细菌DNA提取试剂盒购于上海捷瑞生物工程有限公司;高保真酶PrimeSTAR购自宝生物工程(大连)有限公司;主要仪器有核酸电泳仪(上海天能科技有限公司)、凝胶成像系统Gel DocTMXR+[伯乐生命医学产品(上海)有限公司]、ABI-Veriti梯度PCR仪(美国应用生物系统公司)等。
1.2 方法
1.2.1 大肠埃希菌分离
分离方法采用国家标准GB 4789.38—2012进行,获得纯培养物后通过全自动细菌鉴定系统进一步确认后,使用20%甘油溶液保藏于-80 ℃冰箱中。
1.2.2 基因组DNA提取
在LB平板上划线分离单菌落,37 ℃温度下液体培养10 h,D600在0.6至1.0之间,取500 μL收集菌体,采用上海捷瑞生物细菌基因组抽提试剂盒及操作说明提取基因组DNA,然后进一步通过琼脂糖凝胶电泳确认质量。
1.2.3 PCR扩增
MLST扩增引物adk(adenylate kinase,腺苷酸激酶)、fumC(fumarate hydratase,延胡索酸水合酶)、gyrB(DNA gyrase,DNA解旋酶)、icd(isocitrate/isopropylmalate dehydrogenase,异柠檬酸脱氢酶)、mdh(malate dehydrogenase,苹果酸脱氢酶)、purA(adenylosuccinate dehydrogenase,腺苷酸琥珀酸脱氢酶)和recA(recombinase A,重组酶A) 参考http://mlst.warwick.ac.uk/mlst/dbs/Ecoli/documents/primersColi_html网站中公布的序列委托上海捷瑞生物工程有限公司合成(表1)。50 μL反应体系,包含0.5 μL PrimeSTAR HS DNA 聚合酶、上下游引物(10 μmol·L-1)各1.0 μL、dNTP(2.5 mmol·L-1)4.0 μL和基因组DNA模板0.5 μL。扩增条件:98 ℃ 10 s、57 ℃ 30 s、72 ℃ 1 min,35个循环。扩增完毕后,取PCR产物3 μL通过琼脂糖凝胶电泳进行检测。
1.2.4 测序及MLST分型分析
扩增的单一条带与预计大小相符后,割胶回收后委托杭州擎科梓熙生物技术有限公司进行sanger法测序,测序的峰图文件使用phred/phrap/consed工具包进行拼接、质量验证。拼接获得的保守基因部分序列使用在线工具http://mlst.warwick.ac.uk/mlst/dbs/Ecoli进行分析,获得各看家基因的等位基因编码(allele number),并生成各菌株的ST序列型。
1.2.5 进化树分析
将测得的不同等位基因序列分别通过Clustal进行对位分析,7个基因串联后采用NJ(neighbor-joining)法进行bootstrap值为1 000次的重复构建,使用MEGA6软件进行系统发育树图形绘制。
表1 本试验所需引物
Table 1 Primer sequences used in this study

基因名称Genename引物序列Primersequence(5'-3')退火温度Annealingtemperature/℃adkF:ATTCTGCTTGGCGCTCCGGG54R:CCGTCAACTTTCGCGTATTTfumCF:TCACAGGTCGCCAGCGCTTC54R:GTACGCAGCGAAAAAGATTCgyrBF:TCGGCGACACGGATGACGGC60R:ATCAGGCCTTCACGCGCATCicdF:ATGGAAAGTAAAGTAGTTGTTCCGGCACA54R:GGACGCAGCAGGATCTGTTmdhF:ATGAAAGTCGCAGTCCTCGGCGCTGCTGGCGG60R:TTAACGAACTCCTGCCCCAGAGCGATATCTTTCTTpurAF:CGCGCTGATGAAAGAGATGA54R:CATACGGTAAGCCACGCAGArecAF:CGCATTCGCTTTACCCTGACC58R:TCGTCGAAATCTACGGACCGGA
1.2.6 耐药谱测定
大肠埃希菌鉴定和药敏试验采用梅里埃微生物全自动药敏系统进行检测。
2 结果与分析
2.1 获得菌株和耐药谱测定
采用1.2.1节方法分离的大肠埃希菌菌株经过VITEK 2 Compact系统再次鉴定后,共计获得59株大肠埃希菌,并同时获得每株菌对18种抗生素的耐药谱,共获得24种耐药谱(表2)。根据表2中统计,所有分离株对18种抗生素全敏感有12株;耐1种药物有12株,其中耐磺胺药SXT(甲氧苄啶)有7株;耐2种药AMP(氨苄西林)和SXT的菌株共计13株,是耐药类型中频率最高的。另外,耐3种药物以上共计20株,耐6种以上抗生素有10株。从以上数据可以看出分离的大肠埃希菌耐药类型多样,其中多重耐药现象普遍。由图1得知,所有的分离株中耐AMP和SXT的比例最高,分别为61%和50%;其次为CFZ(头孢唑林)、CRO(头孢曲松)和CIP(环丙沙星),耐药率都超过了20%。
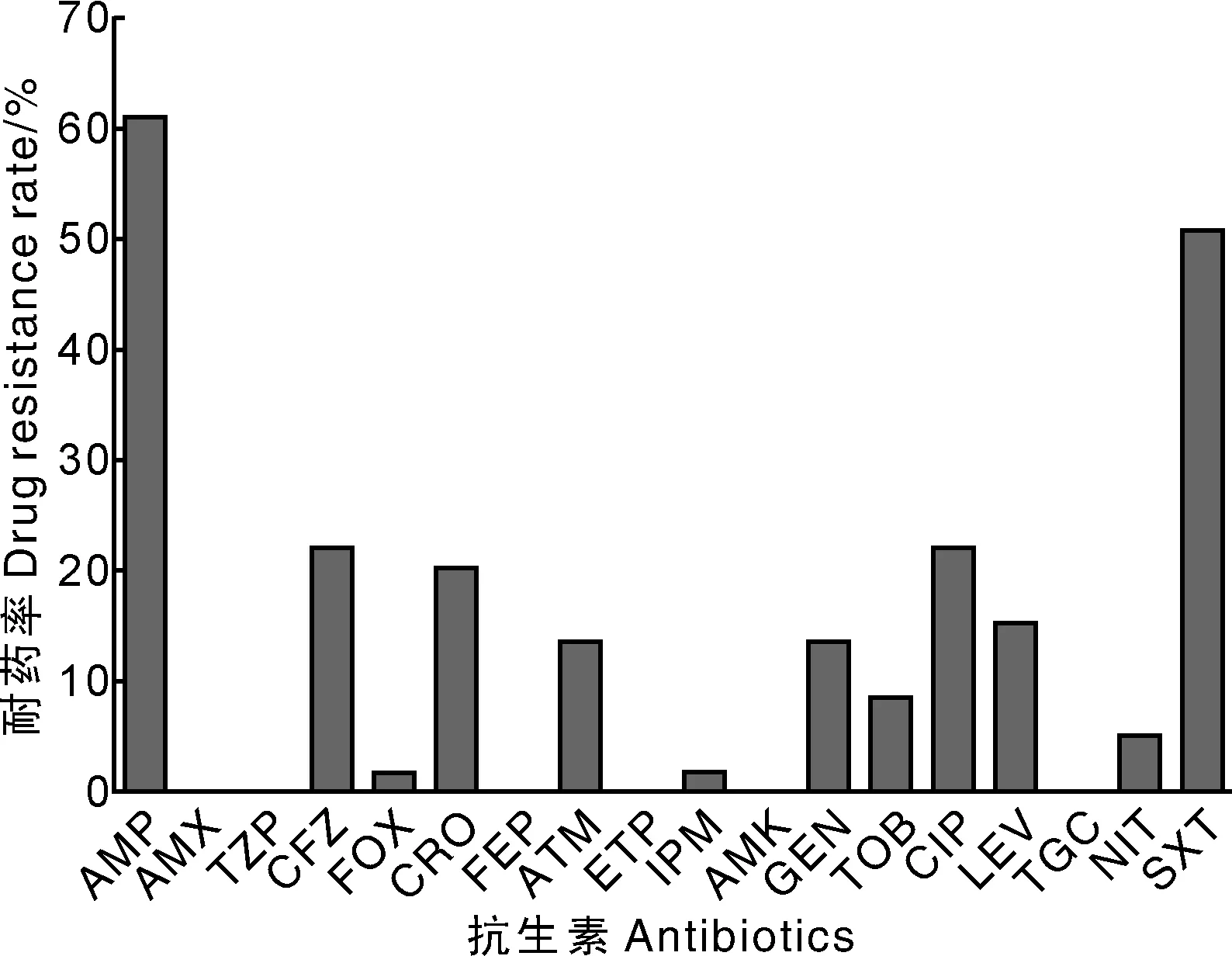
抗生素代码与表2的表注相同The antibiotic code is the same as in Table 2.图1 大肠埃希菌分离株对18种抗生素的耐药率Fig.1 The resistance rates of Escherichia coli to 18 kinds of antibiotics of isolates
2.2 MLST分析
7个看家基因adk、fumC、gyrB、icd、mdh、purA和recA经PCR扩增后电泳均获清晰单一条带(图2),经测序后获得基因的部分保守序列。59组等位基因提交数据库比对后,共计获得38个ST型,另有2种ST型未知,应定义成新的ST型(图3)。这说明分离的大肠埃希菌基因型具有多态性,菌株来源多样化。其中ST-155有7株,ST-10有6株,预示着这两个基因型的大肠埃希菌在3个地区分布较广泛;而其他型均只有1~2株。
2.3 系统发育分析
通过将MLST分型方法使用的7个看家基因测得的序列串联后,对59株大肠埃希菌分离株和大肠埃希菌野生型菌株MG1655进行系统发育AMP(Ampicillin,氨苄西林)、AMX(Amoxicillin,阿莫西林)、TZP(Piperacillin,哌拉西林)、CFZ(Cefazolin,头孢唑林)、FOX(Cefoxitin,头孢西丁)、CRO(Ceftriaxone,头孢曲松)、FEP(Cefepime,头孢吡肟)、ATM(Aztreonam,氨曲南)、ETP(Ertapenem,厄他培南)、IPM(Imipenem,亚胺培南)、AMK(Amikacin,阿米卡星)、GEN(Gentamicin,庆大霉素)、TOB(Tobramycin,妥布霉素)、CIP(Ciprofloxacin,环丙沙星)、LEV(Levofloxacin,左氟沙星)、TGC(Tigecycline,替加环素)、NIT(Nitrofurantoin,呋喃妥因)、SXT(Trimethoprim,甲氧苄啶)。
表2 耐药谱统计
Table 2 Statistics of drug resistance spectrum
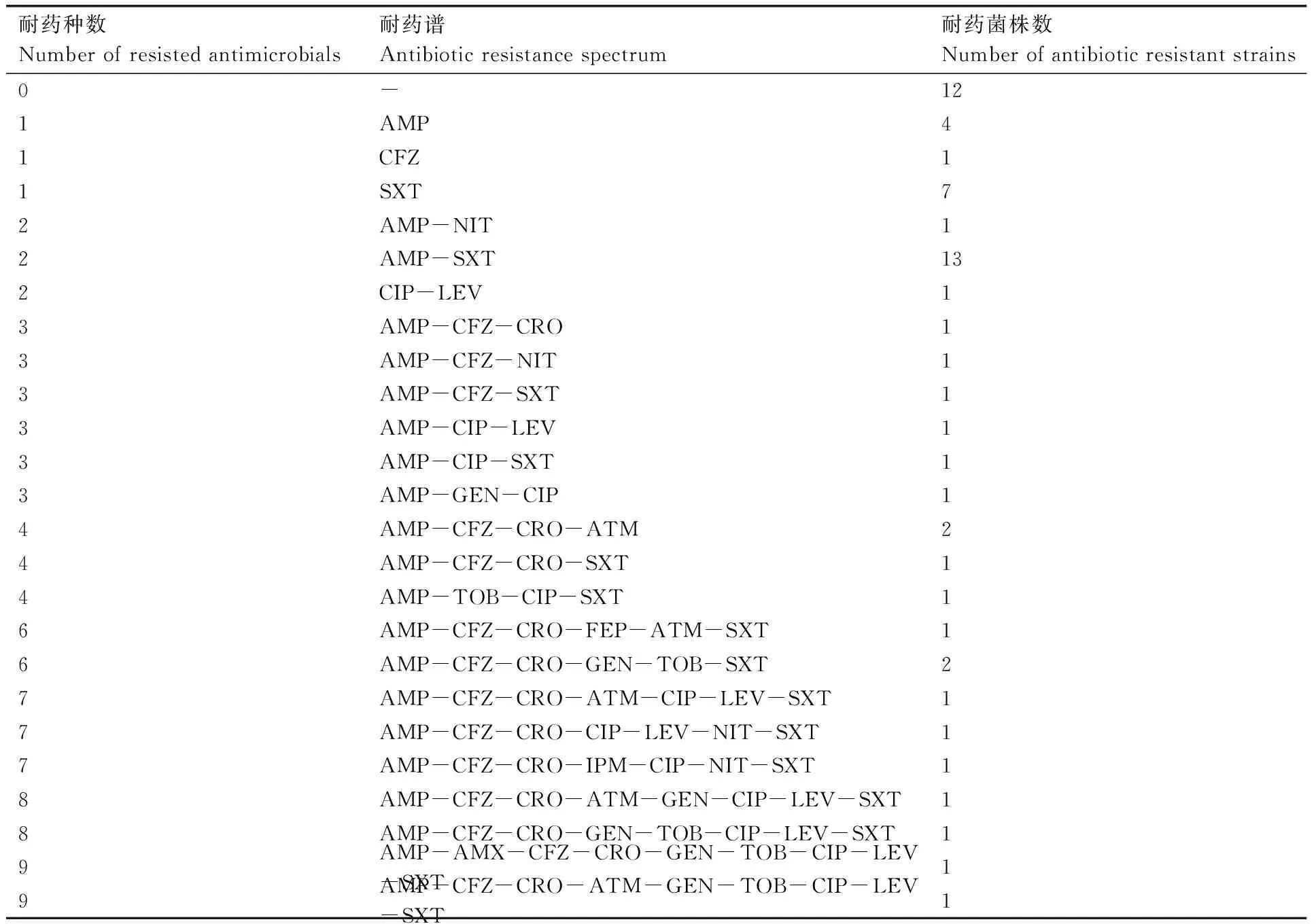
耐药种数Numberofresistedantimicrobials耐药谱Antibioticresistancespectrum耐药菌株数Numberofantibioticresistantstrains0-121AMP41CFZ11SXT72AMP-NIT12AMP-SXT132CIP-LEV13AMP-CFZ-CRO13AMP-CFZ-NIT13AMP-CFZ-SXT13AMP-CIP-LEV13AMP-CIP-SXT13AMP-GEN-CIP14AMP-CFZ-CRO-ATM24AMP-CFZ-CRO-SXT14AMP-TOB-CIP-SXT16AMP-CFZ-CRO-FEP-ATM-SXT16AMP-CFZ-CRO-GEN-TOB-SXT27AMP-CFZ-CRO-ATM-CIP-LEV-SXT17AMP-CFZ-CRO-CIP-LEV-NIT-SXT17AMP-CFZ-CRO-IPM-CIP-NIT-SXT18AMP-CFZ-CRO-ATM-GEN-CIP-LEV-SXT18AMP-CFZ-CRO-GEN-TOB-CIP-LEV-SXT19AMP-AMX-CFZ-CRO-GEN-TOB-CIP-LEV-SXT19AMP-CFZ-CRO-ATM-GEN-TOB-CIP-LEV-SXT1
“-” 代表对抗生素全敏感。The symbol “-” represents all sensitivity to 18 antibiotics.
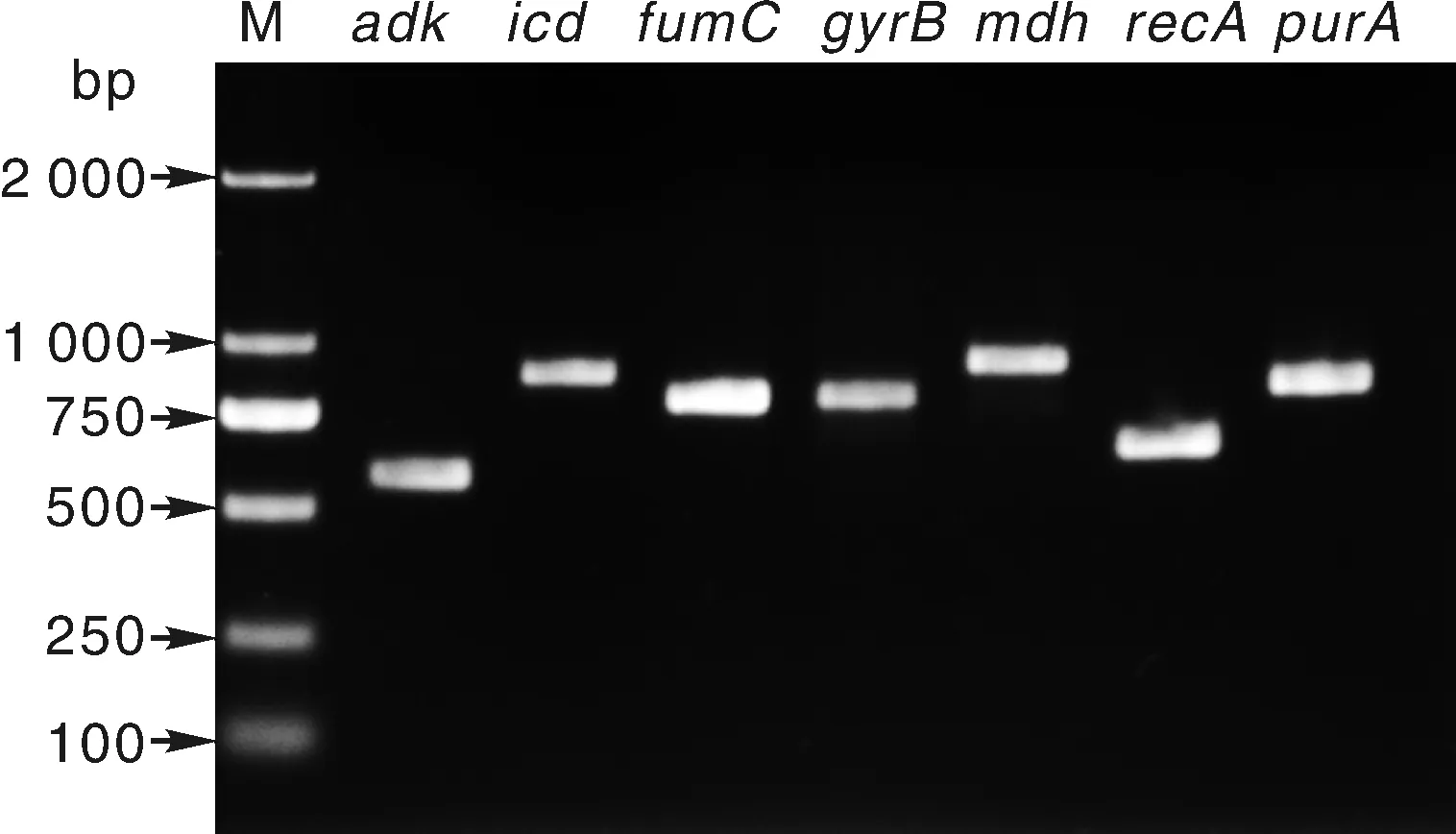
图2 七个看家基因的电泳结果Fig.2 Electrophoresis results of 7 housekeeping genes
树分析(图3)。发现系统发育树有两个明显的分支Group 1 和Group 2,bootstrap值均大于50%,说明分支稳定可信。其中JH市分离的菌株大部分存在于Group 1,少部分存在于Group 2,体现出该地区的特征差异性;HZ和SX市分离株分布没有规律,并且两市分离株同时聚类在Group 1和Group 2中,说明这两个地区间没有地区特征性差异。另外,在该系统发育树中表现出了地区特有分支,例如JH市的ECCJH98-2、ECCJH103-2、ECCJH107-2和ECCJH111-1;SX市的 ECCSX58-2、ECCSX69-2、ECCSX56-1和ECCSX56-2。
3 结论与讨论
大肠埃希菌一直是人和动物的条件致病菌,容易造成人畜共患病[15]。由于畜禽养殖中大量使用抗生素,导致大肠埃希菌多重耐药性越来越严重,直接给人类和环境造成威胁[2]。大肠埃希菌中的耐药基因通常存在于质粒上,而质粒是耐药基因转移的最重要的可移动元件[6,16]。分离的大肠埃希菌即使没有致病性,但是其携带的多重耐药基因质粒对肠杆菌科其他的致病菌或者其他种类病原菌存在非常大的转移风险。冷鲜鸡是禽产品产业链的后端环节,人群可以直接接触,评价其污染的大肠埃希菌的耐药性具有重要卫生意义,对该大肠埃希菌分离株进行基因分型,有助于对这些大肠埃希菌的分布规律进行研究。
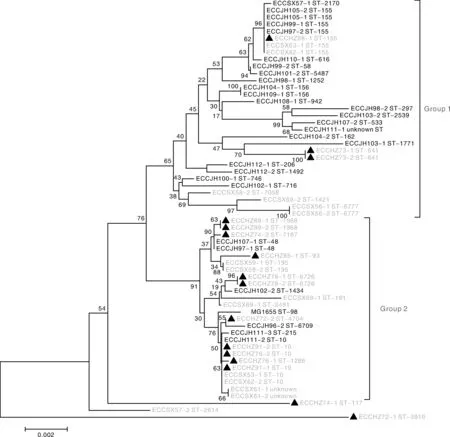
黑体字体为JH市来源的分离株,浅灰色字体为SX市来源的分离株,▲+浅灰色字体为HZ市来源的分离株。The black font strains are isolates from JH city, the grey font strains are isolates from SX city and ▲+ grey font strains are isolates from HZ city.图3 七个看家基因部分序列串联系统发育树Fig.3 A phylogenetic tree of partial sequences of 7 housekeeping genes
本研究从浙江省3个地区市售冷鲜鸡样品中直接分离出大肠埃希菌59株,对于评价其耐药性具有一定的代表性。本研究检测到了24种耐药谱,其中耐AMP-SXT菌株较多,占22.0%。所有菌株中耐药率最高的同样是AMP和SXT,均超过50%,这和养殖过程中这两种药的大量使用相关,两种抗生素耐药基因是否同时存在于同一移动元件上有待进一步研究。通过对分离到的59株大肠埃希菌的7个看家基因进行测序比对,我们分别获得了相应的38种ST型,另外还发现了2种未知类型,反映了分离株的基因型多样性。ST-10和ST-155类型在3个地区均有检出,说明可能污染的来源相同。针对这7个基因序列进一步做系统发育分析后,菌株被分为两个主要的组。我们发现JH市的菌株显示了一定的区域性,而SX和HZ的菌株大多亲缘关系较近,没有明显的区域隔离。该结果可能与冷鲜鸡上游的供应链有关,JH市相对于其他两市距离远,相对独立,冷鲜鸡来源的屠宰场不同,HZ和SX分支距离较近,供应链很可能存在交叉。
本试验通过对以上菌株进行耐药谱测定、MLST分子分型以及系统发育分析,为明确浙江省市售冷鲜鸡污染的大肠埃希菌的耐药状况提供数据支撑。同时为其分子溯源研究提供了思路。
[1] VAN DEN BOGAARD A E, STOBBERINGH E E. Antibiotic usage in animals: impact on bacterial resistance and public health [J].Drugs, 1999, 58(4):589-607.
[2] HAMPTON T. Report reveals scope of US antibiotic resistance threat [J].JAMAtheJournaloftheAmericanMedicalAssociation, 2013, 310(16):1661-1663.
[3] BOTTERY M J, WOOD A J, BROCKHURST M A. Selective conditions for a multidrug resistance plasmid depend on the sociality of antibiotic resistance [J].AntimicrobialAgentsandChemotherapy, 2016, 60(4):2524-2527.
[4] LI J, WANG T, SHAO B, et al. Plasmid-mediated quinolone resistance genes and antibiotic residues in wastewater and soil adjacent to swine feedlots: potential transfer to agricultural lands [J].EnvironmentalHealthPerspectives, 2012, 120(8):1144-1149.
[5] EMBORG H D, BAGGESEN D L, AARESTRUP F M. Ten years of antimicrobial susceptibility testing ofSalmonellafrom Danish pig farms [J].TheJournalofAntimicrobialChemotherapy, 2008, 62(2):360-363.
[6] HU Y, YANG X, LI J, et al. The bacterial mobile resistome transfer network connecting the animal and human microbiomes [J].AppliedandEnvironmentalMicrobiology, 2016, 82(22):6672-6681.
[7] BLAIR J M, WEBBER M A, BAYLAY A J, et al. Molecular mechanisms of antibiotic resistance [J].NatureReviewsMicrobiology, 2015, 13(1):42-51.
[8] ZHOU W, WANG Y, LIN J. Functional cloning and characterization of antibiotic resistance genes from the chicken gut microbiome [J].AppliedandEnvironmentalMicrobiology, 2012, 78(8):3028-3032.
[9] GYLES C L. Antimicrobial resistance in selected bacteria from poultry [J].AnimalHealthResearchReviews, 2008, 9(2):149-158.
[10] WANG Y, ZHANG R, LI J, et al. Comprehensive resistome analysis reveals the prevalence of NDM and MCR-1 in Chinese poultry production [J].NatureMicrobiology, 2017, 2: 16260.
[11] DELGADO-BLAS J F, OVEJERO C M, ABADIA-PATINO L, et al. Coexistence ofmcr-1 andblaNDM-1 inEscherichiacolifrom Venezuela [J].AntimicrobialAgentsandChemotherapy, 2016, 60(10):6356-6358.
[12] LU S, JIN D, WU S, et al. Insights into the evolution of pathogenicity ofEscherichiacolifrom genomic analysis of intestinalE.coliofMarmotahimalayanain Qinghai-Tibet plateau of China [J].EmergingMicrobes&Infections, 2016, 5(12):e122.
[13] WIRTH T, FALUSH D, LAN R, et al. Sex and virulence inEscherichiacoli: an evolutionary perspective [J].MolecularMicrobiology, 2006, 60(5):1136-1151.
[14] WANG Y, TIAN G B, ZHANG R, et al. Prevalence, risk factors, outcomes, and molecular epidemiology of mcr-1-positive Enterobacteriaceae in patients and healthy adults from China: an epidemiological and clinical study [J].TheLancetInfectiousdiseases, 2017, 17(4):390-399.
[15] KOZAK G K, BOERLIN P, JANECKO N, et al. Antimicrobial resistance inEscherichiacoliisolates from swine and wild small mammals in the proximity of swine farms and in natural environments in Ontario, Canada [J].AppliedandEnvironmentalMicrobiology, 2009, 75(3):559-566.
[16] LIU Y Y, WANG Y, WALSH T R, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study [J].TheLancetInfectiousDiseases, 2016, 16(2):161-168.
(责任编辑 卢福庄)
Drug resistance spectrum analysis and MLST genotyping ofEscherichiacolifrom refrigerated chicken in three regions of Zhejiang Province
YANG Hua1,2, HE Xiangxiang1,2,3, XIAO Yingping1,2, QIAN Mingrong1,2, ZHANG Qiaoyan1, TANG Biao1,2,*
(1.InstituteofQualityandStandardforAgro-products,ZhejiangAcademyofAgriculturalSciences,Hangzhou310021,China; 2.StateKeyLaboratoryBreedingBaseforZhejiangSustainablePestandDiseaseControl,ZhejiangAcademyofAgriculturalSciences,Hangzhou310021,China; 3.CollegeofFoodScienceandEngineering,NorthwestA&FUniversity,Yangling712100,China)
Escherichiacoliis a common opportunistic pathogen in food. Its drug resistance is severe in which the related resistance genes can easily spread. In this study, 59 strains ofE.colifrom refrigerated chicken in three regions of Zhejiang Province were isolated. Using drug resistant spectrum determination, 24 types of drug resistant spectrum were found, which showed that the drug resistant types ofE.coliisolates were multifarious. Meanwhile, all theE.coliisolates were analyzed with MLST genotyping. The results showed that a total of 38 ST types were obtained, and two new ST types were found as well. On this basis, the phylogenetic analysis of 7 housekeeping genes showed a certain degree of the regional differences. This research results can provide scientific reference for the evaluation of drug resistance and traceability analysis of avianEscherichiacoliin Zhejiang province, and a wider region in China.
Escherichiacoli; bacterial drug resistance; multilocus sequence typing (MLST); phylogenetic tree
10.3969/j.issn.1004-1524.2017.06.05
2017-05-12
浙江省农业科学院青年人才培养项目(2017R19R08E01);浙江省植物有害生物防控重点实验室——省部共建国家重点实验室培育基地(2011DS700124-ZZ1703);浙江省重点研发项目(2015C02041)
杨华(1972—),男,浙江绍兴人,硕士,高级畜牧师,研究方向为畜产品质量安全。E-mail: yanghua806@hotmail.com
*通信作者,唐标,E-mail: tb_411@163.com
S852.61
A
1004-1524(2017)06-0888-06
