常见遗传性大肠癌的外科治疗
2017-06-19阿地力克然木刘方奇徐烨
阿地力·克然木 刘方奇 徐烨
常见遗传性大肠癌的外科治疗
阿地力·克然木 刘方奇 徐烨
大肠癌在我国的发病率和死亡率均呈逐年上升趋势,其中5~6%为遗传性大肠癌[1]。遗传性大肠癌具有遗传性、家族聚集性、高发性、多发性和肿瘤发生的多器官性等特点,对人类的生存构成极大的威胁。家族中突变基因携带者患相关肿瘤风险显著高于普通人群,早期筛查及干预可降低其患癌风险。本文结合常见遗传性大肠癌(Lynch综合征和家族性腺瘤性息肉病)的临床及遗传学特征、基因检测等内容阐述其外科治疗,以期为医务人员治疗的选择提供依据,采取精准的干预及筛查措施,降低患者患癌风险。
结直肠肿瘤; 外科手术; 遗传性大肠癌; Lynch 综合征; 家族性腺瘤性息肉病; 精准医疗
遗传性大肠癌大体分为息肉病性综合征和非息肉病性综合征,前者可分为腺瘤性息肉病综合征和错构瘤息肉病综合征两类。腺瘤性息肉病综合征包括家族性腺瘤性息肉病(familial adenomatous polyposis,FAP)及其亚型,错构瘤息肉病综合征包括遗传性色素沉着-消化系息肉病综合征(peutz-jeghers syndrome,PJS)、家族性幼年性结肠息肉病、PTEN错构瘤肿瘤综合征(PTEN hamartoma tumor syndrome,PHTS)、遗传性混合息肉病综合征(hereditary mixed polyposis syndrome,HMPS)等一系列疾病[2]。后者又分为Lynch综合征(Lynch syndrome,LS)[3]和家族性癌症综合征。Lynch 综合征(3~5%)和家族性腺瘤性息肉病(familial adenomatous polyposis,FAP)(0~1%)是最常见的遗传性大肠癌。由于遗传病因特殊、临床病理特点突出,遗传性大肠癌是目前临床肿瘤学研究的热点。本文着重介绍FAP和Lynch综合征的外科处理以及当前精准医疗时代的背景下如何结合基因突变检查结果为患者提供更为精准的外科治疗。
一、 家族性腺瘤性息肉病(FAP)
(一)FAP的发病原因及临床表型
FAP是APC基因突变引起的常染色体显性遗传病[4],约占大肠癌的1%[5-6]。FAP由APC基因突变造成,该基因位于常染色体5p 22.2。超过80%的FAP患者可检测到APC基因突变[5-7]。典型的FAP的定义为:以遍布整个大肠、数目超过100个以上的腺瘤性息肉和微腺瘤为临床表现的常染色体显性遗传综合征。患者十几岁时开始出现腺瘤,如不治疗,至40岁时100%的患者会转变为结直肠癌[8]。依据遗传病因和临床表型的不同[9],FAP又可分为经典型家族性腺瘤性息肉病(classical FAP,CFAP)、衰减型家族性腺瘤性息肉病(attenuated FAP,AFAP)(CFAP和AFAP临床特征的区别见表1)、MYH相关性息肉病(MYH-associated polyposis,MAP)、Gardner综合征(Gardner syndrome,GS)、Turcot综合征(Turcot syndrome,TS)等亚型[2]。
(二)FAP的外科治疗
1.FAP术式
各种FAP临床亚型的共同特征就是结直肠腺瘤性息肉,由于其恶变率高,因此目前临床上对于FAP的结直肠腺瘤性息肉仍主采取外科手术治疗。FAP的手术方式大致有三类[2,13-14]:全结肠切除联合回肠末端造瘘术(total proctocolectomy and permanent ileostomy,TPC)、全结直肠切除联合回肠储袋肛管吻合术(ileal pouch-anal anastomosis,IPAA)和全结肠切除联合回肠直肠吻合术(total colectomy and ileorectal anastomosis,IRA)。三种术式的优缺点对比如表2所示。对于TPC来讲,这是最有希望治愈FAP的外科术式,是传统的经典手术,以前被认为该术式彻底性最佳,无直肠病变复发和癌变之虑,但此术式带来的并发症多,永久造瘘带来诸多不便,加以盆腔内解剖易损伤神经而影响性功能和生育功能,对年轻人实属不宜。因此,它通常被保留作为局部晚期直肠癌或者失去肛门括约肌功能的患者的选择,但目前此方法已较少使用,故目前主要采用的术式是IPAA和IRA。对于IRA来讲,手术时保留7~10 cm直肠,全切其余结直肠,行回肠直肠吻合。本方法手术损伤小、保留了排便、排尿和性功能,并发症低。但残留的直肠仍有癌变危险,须定期随诊,对发现的息肉及时进行电灼或手术切除。此术式适用于青少年患者或高龄患者、肠道息肉少的患者。对于IPAA来讲,本手术可保留肛门,肛门功能尚可,排便功能相对较好,但手术复杂,并发症较多,影响患者性功能和生育功能。随着手术技术的改进,吻合器械的使用,降低了并发症,使IPAA手术变得简单安全,已成为治疗FAP越来越普遍的方法。
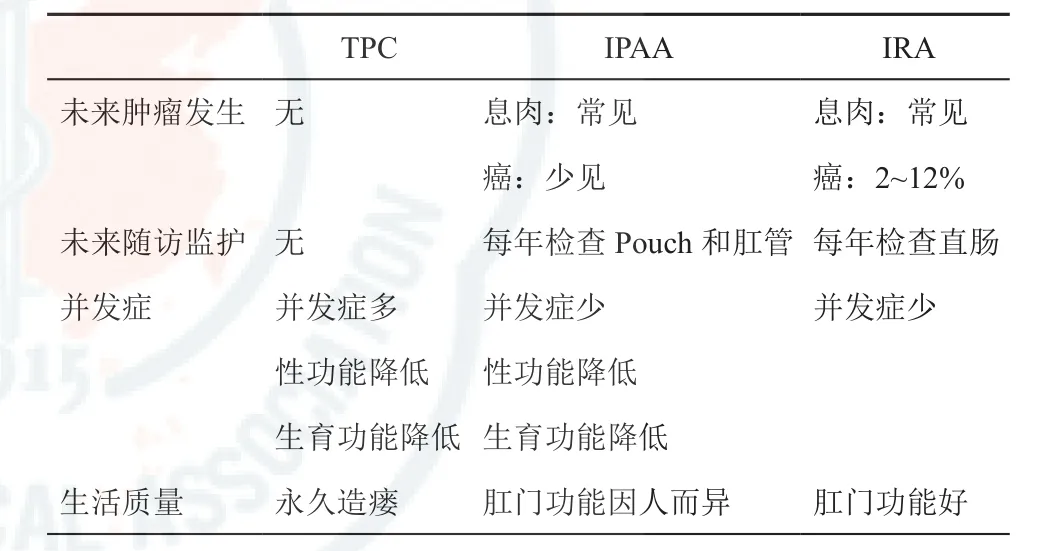
表2 FAP各种术式的优缺点对比
2.FAP外科治疗的选择
对于FAP患者是否手术,何时手术以及手术方式的选择需要考量发病年龄、纤维瘤病、直肠腺瘤数目、患者意愿、生育愿望等多种因素。
国外一项[15]26个FAP登记中心合作的研究约6 600个FAP患者发现仅14例患者发生癌变的年龄小于20岁,约占0.2%,其中:1例患者9岁,其他癌变均发生在17~19岁,且这些患者腺瘤负荷大或者有症状。该研究提示青少年FAP发生癌变的可能性低。而另外一项研究[16]统计多中心共1 073大肠癌患者提示11~15岁的大肠癌患者只有2例(占0.2%),16~20岁的有15例(占1.3%),这也提示小于20岁患者癌变率低。由此得知,对于青少年FAP患者的治疗除非腺瘤负荷大或者有明显的临床症状,否则可以选择肠镜监测,使用西乐葆等选择性环氧化酶-2抑制剂控制和减少息肉负荷,延迟外科治疗。
纤维瘤病是FAP患者除了结直肠癌之外主要的死亡原因。芬兰一项回顾性研究[17]202名FAP患者发现患纤维瘤病的一组之前接受肠切除年龄(28.6岁)比没有患纤维瘤病的一组(34.5岁)要早(P<0.05),得出对FAP患者较早行预防性肠切除可能增加FAP患者患纤维瘤病的风险。为了验证这一点,Durno等[18]回顾性分析930名FAP患者发现121名患者(14%)出现纤维瘤病,女性比男性更容易得纤维瘤病(分别为17%和13%,P<0.03),女性患者中18岁之前接受肠切除的患者患纤维瘤病的风险比18岁之后接受手术高2倍(P<0.01),而男性患者却没有明显变化。18岁之前接受肠切除的女性患者患纤维瘤病的风险比较晚行手术的男性高2.5倍,指出女性FAP患者比男性患者更易患纤维瘤病,女性较早行肠切除明显增加患纤维瘤病的风险。因此对于女性FAP无腺瘤癌变者应适当延迟手术以降低纤维瘤病的风险,对于小于18岁的女性FAP无腺瘤癌变者则可推迟至18岁之后再选择手术治疗。
术前直肠腺瘤数目被认为是FAP患者选择何种术式的因素之一,Church等[19]为了研究术前直肠镜对FAP严重性评估的可靠程度以及患者行IRA术后的预后效果,回顾性分析213名FAP患者,得出如表3的结果,指出术前直肠镜检测腺瘤数目是术前评估FAP患者严重性程度及预后的有效手段。因此,术前对于直肠腺瘤数目小于20枚的FAP患者适宜选择IRA,而大于20枚的患者可能需要IPAA或者TAC,以便降低二次直肠切除的风险。

表3 直肠腺瘤数与IRA术后直肠癌变率[19]
为了研究IRA和IPAA对FAP患者生育功能的影响,Olsen等[20]回顾性分析162例FAP女性患者,将其分为未行手术者、行IRA手术者、行IPAA手术者三组,比较手术前后怀孕时间间隔,发现前两组相比生育能力无差别,一组、三组对比患者生育能力下降50%。而另外几个回顾性研究[21-23]提示接受IPAA术式的男性患者的射精障碍率和阳痿率分别达16%和25%,明显高于IRA患者的出现的生育功能障碍(3%和1%)。IPAA会影响FAP患者的生育功能,因此对于育龄期或者有生育愿望的患者选择外科治疗方式时一定要考虑IPAA手术对生育功能的影响,当直肠腺瘤数目小于20枚时可以选择影响较小的IRA术式。
二、 Lynch 综合征
(一)Lynch 综合征的临床特点
Lynch 综合征是一种常染色体显性遗传病是与DNA错配修复基因(mismatch repair,MMR)突变有关,可累及多个脏器的遗传性癌症综合征。50~80%的Lynch 综合征患者会发生结直肠癌,约占结直肠癌的5~10%[24]。Lynch综合征有以下特点:发病年龄早(中位年龄约44岁,较散发性结直肠癌提前约20年);近侧大肠癌多见(约70%位于脾曲近侧);同时或异时多原发大肠癌发生率高(结肠不全切除后10年内约40%再发);结直肠外恶性肿瘤发生率高(包括子宫内膜癌、卵巢癌、胃癌、小肠癌及泌尿系统癌等)。
(二)Lynch综合征的外科处理
在临床上遇到的Lynch综合征患者分为4种[25]:(1)已经确诊的Lynch综合征患者;(2)行大肠癌手术,术后确诊为Lynch综合征;(3)行妇科或其他器官相关癌症手术后确诊为Lynch综合征;(4)基因检测发现MMR突变携带者。
对术前已经确诊的Lynch综合征患者,应行预防性结肠次全切除或全结直肠切除,也有专家认为行患病区域的标准根治术,即肠段切除。对于打算行预防性结肠次全切除或全结直肠切除的患者,如肿瘤位于结肠,则行IRA,术后终身对直肠行肿瘤筛查;如在直肠则行IPAA。Haanstra等[26]的临床研究得出排便功能上次全切除术后比在部分肠段切除更差,但在Lynch综合征的两种类型(肠段切除和全结肠切除)的手术后,生活质量没有差异。而Kalady等[27]回顾性研究296例符合Amsterdam标准的患者发现253例肠段切除的患者中74例(33%)检测到异时多原发腺瘤,其中高危腺瘤48例(22%),55位患者(25%)发生异时多原发癌(平均间隔时间5.7年)。相比之下,接受IRA的43例患者中有4例(11%)发生异时多原发腺瘤,3例(8%)发生异时多原发癌(平均间隔时间18年)。接受肠段切除的患者具有高的异时性高风险腺瘤和癌的发生率。Cirillo等[28]的回顾性研究发现异时性结直肠癌患者的死亡风险增加了6倍且异时性癌的分期比原发肿瘤晚,直肠癌的一级家族史与直肠癌的风险增加有关。因此,对于任何手术选择,内窥镜监测是必要的,临床医生与患者讨论术式的选择时,应该强调两种外科手术的所有优点和缺点,包括生活质量和功能结果[26]。对于术后确诊的Lynch综合征患者,要定期进行结肠镜检查,发现有腺瘤癌变倾向时及早进行根治性手术,防止癌变发生。对于MMR突变携带者,应进行严密的监测。
三、精准医疗时代遗传性大肠癌的治疗
精准医疗因其精准性和便捷性开启了医学新时代,基因测序可以我们找出癌症的突变基因,从而帮助我们早期筛查及干预,降低家族中突变基因携带者患癌风险。
(一)FAP的基因型、表型与治疗的关系
FAP由APC基因突变造成,而临床表型与APC基因突变位点相关[2,29-30]:密码子169~1 393 之间突变表现为CFAP;在此区域5’端(密码子1 157)或3’端(密码子1 595~2 843)突变表现为AFAP,结直肠息肉数目通常少于100枚,AFAP患者发病年龄一般在30~50岁,癌变率为69%;密码子1 250~1 464 之间突变的表现为严重型CFAP,结直肠息肉最多数量可达几千枚,一般在20岁之前发病,癌变率为100%,对于该区域突变的患者青少年期开始应检测和及时诊断,及时选择外科治疗;密码子463~1 444 之间突变常伴发视网膜病变;密码子1 445~1 578 之间突变与伴发硬纤维瘤、骨瘤、表皮囊肿有关,根据之前所述,纤维瘤病是FAP患者第二死亡原因,对于该区域突变的患者一定要权衡手术时间的选择,降低纤维瘤的风险;位于密码子279~1 309之间突变者与十二指肠腺瘤相关,对于该区域突变的患者需定期胃镜监测十二指肠。FAP基因型与IRA术后直肠二次手术也有关系,国外报道[31]一项多个国家息肉登记中心研究475例行TAC/IRA手术的且基因型已知患者,根据临床表型分为衰减型,中间型和严重型,结果发现IRA术后二次手术的累积风险度分别为10%,39%和61%(P<0.05),初次全结肠切除术后直肠癌变率的累积风险分别为3.7%,9.3%和8.3%组(P≤0.05),故对于中间型和严重型患者预防性手术时IPAA或者TPC术式可能会降低二次手术风险。结直肠癌和纤维瘤病是FAP患者死亡的主要原因,而对FAP行基因筛查和诊断可能帮助医务人员监测和诊断FAP患者,并且选择正确的外科治疗,降低患者患癌风险。
(二)Lynch综合征的基因型、表型与治疗的关系
Lynch综合征同时或异时性发生肠内外肿瘤的风险度高,包括子宫内膜癌、卵巢癌、胃癌等一系列相关肿瘤。随着基因检测的普及和深入,Lynch综合征的致病基因MMR逐渐被阐明,目前国际上报道的主要有MLH1,MSH2,MSH6,PMS2以及EPCAM共5个,不同的基因突变型其临床表型亦不同[2,33-34](见表5)。MLH1突变者主要以结直肠肿瘤为主,因此对于结直肠有症状的患者要行相应的外科治疗,对于没有症状的患者要肠镜定期监测。MLH1突变者肠外肿瘤的发生率少于MSH2,MSH2突变者更易出现肠外肿瘤,故需定期监测胃肠道、子宫等常见肠外肿瘤发生部位;MSH2/MSH6突变女性患者子宫内膜癌的患病风险高,故对于年龄在30~35岁的女性患者应每年或者每两年进行一次子宫内膜活检。并且对于已完成生育的年龄大于35岁的患者可行预防性的子宫及双侧输卵管、卵巢切除[35]。基因检测和筛查可以帮助医护人员随访监测肠道及肠外恶性肿瘤,适时选择正确的外科治疗手段,降低和预防患者及高危家系的患癌风险。
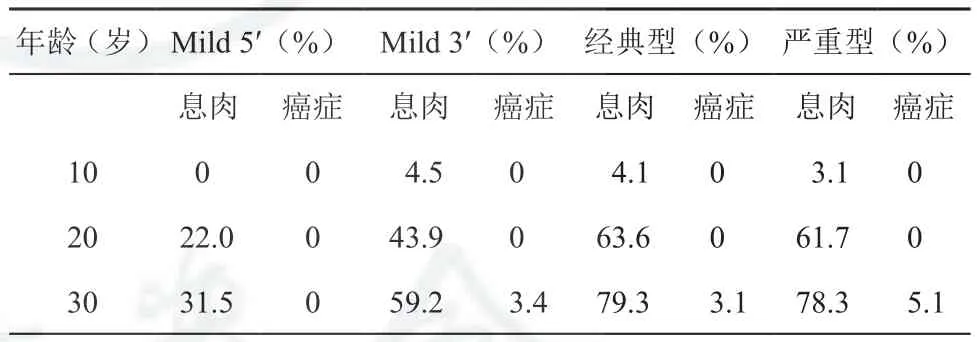
表4 突变表型与癌变率[32]

表5 不同基因突变型及临床表型[2,33-34]
目前二代测序应用技术广泛应用于遗传性大肠癌基因的检测,由于该项技术可同时进行上百万个DNA片段的测序,因此测序成本和时间都大大减少,解决了以往诊断和筛查工作中遇到诸多难题,带来了极大的方便。二代测序已经开始运用于遗传性大肠癌高危家系相关基因的检测中,随着深入研究,一些未知的遗传相关基因也开始被发现和研究。尽管二代测序拥有高通量、高灵敏度、自动化程度高等突出优势,但与一代测序相比,对于序列已知的单基因的突变检测,工作量仍较大,费用偏高。基于我国国情,二代测序工作尚处于起步阶段,很多医院由于缺少相关专业人员、物力等制约着二代测序的推广和肿瘤筛查工作,更为重要的是二代测序测出来的很多遗传基因目前我们无法确定是否为有意义的变异,如何解读这些数据还需多学科团队共同努力。相信随着社会的发展,专业人员及多学科团队的完善,能够制定属于我国国情的遗传性大肠癌的肿瘤筛查标准,基因检测流程和监测随访治疗规范。
[ 1 ] 李晓芬, 袁瑛, 张苏展. 中国人遗传性大肠癌综合征的特征及诊疗规范 [J]. 中国癌症杂志, 2015, 25(11): 841-848.
[ 2 ] 蔡三军. 循证结直肠肛管肿瘤学 [M]. 上海: 上海科学技术出版社, 2016: 650.
[ 3 ] Lynch HT, Riley BD, Weissman SM, et al. Hereditary nonpolyposis colorectal carcinoma (HNPCC) and HNPCC-like families: Problems in diagnosis, surveillance, and management [J]. Cancer, 2004, 100(1): 53-64.
[ 4 ] Wachsmannova-Matelova, Stevurkova V, Adamcikova Z, et al. Different phenotype manifestation of familial adenomatous polyposis in families with APC mutation at codon 1309 [J]. Neoplasma, 2009, 56(6): 486-489.
[ 5 ] Baglioni S, Genuardi M. Simple and complex genetics of colorectal cancer susceptibility [J]. Am J Med Genet C Semin Med Genet, 2004, 129C(1): 35-43.
[ 6 ] Jo WS, Chung DC. Genetics of hereditary colorectal cancer [J]. Semin Oncol, 2005, 32(1): 11-23.
[ 7 ] Vasen, Bulow. Guidelines for the surveillance and management of familial adenomatous polyposis (FAP): a world wide survey among 41 registries [J]. Colorectal Dis, 1999, 1(4): 214-221.
[ 8 ] Jo WS, Chung DC. Genetics of hereditary colorectal cancer [J]. Semin Oncol, 2005, 32(1): 11-23.
[ 9 ] Rustgi AK. The genetics of hereditary colon cancer [J]. Genes Dev, 2007, 21(20): 2525-2538.
[ 10 ] Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature [J]. Crit Rev Oncol Hematol, 2007, 61(2): 153-161.
[ 11 ] Sieber OM, Tomlinson IP, Lamlum H. The adenomatous polyposis coli (APC) tumour suppressor--genetics, function and disease [J]. Mol Med Today, 2000, 6(12): 462-469.
[ 12 ] Knudsen AL, Bisgaard ML, Bulow S. Attenuated familial adenomatous polyposis (AFAP). A review of the literature [J]. Fam Cancer, 2003, 2(1): 43-55.
[ 13 ] Wuthrich P, Gervaz P, Ambrosetti P, et al. Functional outcome and quality of life after restorative proctocolectomy and ileo-anal pouch anastomosis [J]. Swiss Med Wkly, 2009, 139(13-14): 193-197.
[ 14 ] Edlich R, Cross CL, Wack CA, et al. Revolutionary advances in the diagnosis and treatment of Familial Adenomatous Polyposis [J]. J Environ Pathol Toxicol Oncol, 2009, 28(1): 47-52.
[ 15 ] Church JM, McGannon E, Burke C, et al. Teenagers with familial adenomatous polyposis: what is their risk for colorectal cancer? [J]. Dis Colon Rectum, 2002, 45(7): 887-889.
[ 16 ] Vasen HF, Moslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP) [J]. Gut, 2008, 57(5): 704-713.
[ 17 ] Heiskanen I, Jarvinen HJ. Occurrence of desmoid tumours in familial adenomatous polyposis and results of treatment [J]. Int J Colorectal Dis, 1996, 11(4): 157-162.
[ 18 ] Durno C, Monga N, Bapat B, et al. Does early colectomy increase desmoid risk in familial adenomatous polyposis? [J]. Clin Gastroenterol Hepatol, 2007, 5(10): 1190-1194.
[ 19 ] Church J, Burke C, McGannon E, et al. Predicting polyposis severity by proctoscopy: how reliable is it? [J]. Dis Colon Rectum, 2001, 44(9): 1249-1254.
[ 20 ] Olsen KO, Juul S, Bulow S, et al. Female fecundity before and after operation for familial adenomatous polyposis [J]. Br J Surg, 2003, 90(2): 227-231.
[ 21 ] Bissett IP, Hill GL. Extrafascial excision of the rectum for cancer: a technique for the avoidance of the complications of rectal mobilization [J]. Semin Surg Oncol, 2000, 18(3): 207-215.
[ 22 ] Meagher AP, Farouk R, Dozois RR, et al. J ileal pouch-anal anastomosis for chronic ulcerative colitis: complications and longterm outcome in 1310 patients [J]. Br J Surg, 1998, 85(6): 800-803.
[ 23 ] Havenga K, Maas CP, DeRuiter MC, et al. Avoiding long-term disturbance to bladder and sexual function in pelvic surgery, particularly with rectal cancer [J]. Semin Surg Oncol, 2000, 18(3): 235-243.
[ 24 ] 徐烨, 蔡三军, 莫善兢, 等. 遗传性非腺瘤病性结直肠癌的临床特征与诊断原则——附22个家族的报告. 中国广东广州: 2002: 4. [ 25 ] 杨继清, 刘新良, 邵永孚. Lynch综合征诊治进展 [J]. 河北医药, 2013, 35(14): 2185-2186.
[ 26 ] Haanstra JF, de Vos TNCW, Gopie JP, et al. Quality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomy [J]. Dis Colon Rectum, 2012, 55(6): 653-659.
[ 27 ] Kalady MF, Mc Gannon E, Vogel JD, et al. Risk of colorectal adenoma and carcinoma after colectomy for colorectal cancer in patients meeting Amsterdam criteria [J]. Ann Surg, 2010, 252(3): 507-511, 511-513.
[ 28 ] Cirillo L, Urso ED, Parrinello G, et al. High risk of rectal cancer and of metachronous colorectal cancer in probands of families fulf i lling the Amsterdam criteria [J]. Ann Surg, 2013, 257(5): 900-904.
[ 29 ] Soravia C, Berk T, Madlensky L, et al. Genotype-phenotype correlations in attenuated adenomatous polyposis coli [J]. Am J Hum Genet, 1998, 62(6): 1290-1301.
[ 30 ] 于恩达, 徐晓东, 孟荣贵. 家族性腺瘤性息肉病的临床特点及研究现状 [J]. 第二军医大学学报, 2006, 27(4): 349-352.
[ 31 ] Nieuwenhuis MH, Bulow S, Bjork J, et al. Genotype predicting phenotype in familial adenomatous polyposis: a practical application to the choice of surgery [J]. Dis Colon Rectum, 2009, 52(7): 1259-1263.
[ 32 ] Newton KF, Mallinson EK, Bowen J, et al. Genotype-phenotypecorrelation in colorectal polyposis [J]. Clin Genet, 2012, 81(6): 521-531.
[ 33 ] Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer [J]. Cell, 1993, 75(6): 1215-1225.
[ 34 ] Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer [J]. Nature, 1994, 368(6468): 258-261.
[ 35 ] 刘大江, 杜文静.Lynch综合征相关子宫内膜癌新进展[J].国际妇产科学杂志, 2016, 43(1):67-69.
The surgical treatment of common hereditary colorectal cancer
Adili Keranmu, Liu Fangqi, Xu Ye.Department of Colorectal Surgery, Fudan University Shanghai Cancer Center & Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China
Xu Ye, Emai: xuye021@163.com
The incidence and mortality of colorectal cancer in China are increasing year by year, of which 5% to 6% of are hereditary colorectal cancer. Hereditary colorectal cancer has the characteristics of hereditary, familial aggregation, high incidence, multiple incidence and multiple organ tumors and so on, which threaten our life and lead to death. Family of mutation carriers are at signif i cantly greater risk of developing colorectal cancer than the general population. Early screening and intervention can reduce the risk of cancer. Here we summarize the clinical and genetic characteristics of hereditary colorectal cancer syndrome (Lynch Syndrome and Familial Adenomatous Polyposis), and overview the surgical treatment, genetic testing and screening so as to provide the basis for surgeons choosing the medical methods to take accurate intervention and screening measures and reduce the risk of cancer.
Colorectal neoplasms; Surgical procedures, operative; Hereditary colorectal cancer; Lynch syndrome; Familial adenomatous polyposis; Precision medicine
2016-09-13)
(本文编辑:杨明)
10.3877/cma.j.issn.2095-3224.2017.03.015
国家自然科学基金面上项目(No.81472620);上海市卫生系统重要疾病联合公关项目(重大项目)(No.2014ZYJB0101);上海市“科技创新行动计划”实验动物研究领域科技支撑项目(No.13140902100)
200032 上海,复旦大学附属肿瘤医院大肠外科;复旦大学上海医学院肿瘤学系
徐烨,Email:xuye021@163.com
阿地力·克然木, 刘方奇, 徐烨.常见遗传性大肠癌的外科治疗[J/CD].中华结直肠疾病电子杂志, 2017, 6(3): 243-248.
