前列腺癌基因外显子区突变位点筛查及与前列腺癌易感性的关系
2017-06-05糜远源吴升祝黎洁
糜远源,吴升,祝黎洁
(南通大学第三附属医院暨无锡市第三人民医院,江苏无锡214041)
前列腺癌基因外显子区突变位点筛查及与前列腺癌易感性的关系
糜远源,吴升,祝黎洁
(南通大学第三附属医院暨无锡市第三人民医院,江苏无锡214041)
目的 筛查前列腺癌(PCA)基因外显子区的突变位点,并分析其与PCA易感性的关系。方法 6例PCA患者,留取PCA组织和癌旁正常组织标本。采用液相杂交捕获技术分析PCA、癌旁正常组织全基因外显子区的单核苷酸多态性(SNPs),采用Illumina HiSeq 2000平台高通量测序将捕获富集并验证的PCA、癌旁正常组织基因外显子区SNPs的DNA文库进行突变位点比对,筛选PCA基因外显子区突变位点。采用bwa软件与参考基因组hg19作比对,分析PCA外显子区功能性突变位点的生物信息学信息,分析功能性突变位点与PCA易感性的关系。结果 共发现4个未报道的功能性SNPs突变位点,分别为BRCA1 rs16941、MET rs45483396、P53 rs121912655、COPB2 rs7374710。进一步通过临床样本检测结果显示,BRCA1 rs16941与COPB2 rs7374710两个SNP不仅均与PCA易感性相关(OR 分别为 1.47,1.27;95% CI 分别为 1.14~1.72;1.20~1.41),而且与PCA患者PSA值 及Gleason评分均呈正相关(OR分别为1.25,1.37;95%CI= 1.10~1.84,1.20~2.34;P均<0.01)。结论 PCA外显子区存在BRCA1 rs16941、COPB2 rs7374710功能性突变位点,携带有BRCA1 rs16941、COPB2 rs7374710突变位点的个体罹患PCA的可能性较高。
前列腺肿瘤;前列腺癌;液相杂交捕获技术;基因突变;外显子;单核苷酸多态性
2016年最新流行病学数据显示前列腺癌(PCA)仍是美国男性最常见的恶性肿瘤,病死率仅次于肺癌[1]。我国PCA发病率及病死率正逐年升高[2,3]。早期筛查和治疗是一直是PCA研究的热点。目前外周血前列腺特异抗原(PSA)是临床最常用的筛查PCA的指标,虽其诊断PCA复发的敏感度较高,PSA 4 ng/mL阈值诊断PCA的敏感性、特异度均较低[4]。可能原因为前列腺炎症、良性前列腺增生(BPH)及感染时均可出现外周血PSA水平升高[5~7]。单核苷酸多态性(SNPs)是指人群中2个相配对基因DNA序列的某一位点出现的差异和多态性,常常用于疾病相关遗传基因的定位、克隆、鉴定、人群易感性和疾病发生进展的预测[8,9]。目前为止已发现50多个SNPs与高加索人群PCA的发病相关[10];中国人群中一发现的PCA易感性SNPs有二十多种,最为特异的是9q31.2(rs817826)和19q13.4(rs103294)[11]。我们采用液相杂交捕获技术筛查PCA基因外显子区的突变位点,旨在发现未报道过的功能性突变位点并分析其与PCA易感性的关系。现将结果报告如下。
1 材料与方法
1.1 临床资料 收集2013年8月~2016年2月间南通大学第三附属医院收治的PCA患者156例,年龄51~75(65.2±7.4)岁。所有患者均经前列腺穿刺活检和PCA根治手术病理检查证实,并经病史、临床症状和体征及其他辅助检查(如X线、CT、MR等)排除其他类型恶性肿瘤。术中留取PCA组织及癌旁正常组织标本,-80 ℃保存备用。本研究通过南通大学第三附属医院伦理委员会审核通过,患者及其家属均填写知情同意书并备案。
1.2 材料 石蜡包埋组织DNA提取试剂盒 (中国Tiangen公司;DP331),Gnomegen DNA测序文库制备试剂盒(美国Gnomegen公司;K02422),Gnome size selector(美国Gnomegen公司;R02424L),PCR仪(美国ABI公司;2720),Concentrator plus 真空离心浓缩仪(德国eppendorf公司),小型高速离心机(德国eppendorf公司,5418),实时定量PCR仪(德国eppendorf公司,4376600 ),通用电泳仪(杭州汇尔仪器, JY300C),涡旋仪(北京京辉凯业科技有限公司;JHX24H),金属浴(杭州瑞诚仪器有限公司;DH100-1),250 bp DNA Marker (中国捷瑞公司,DL250+,100T),PrimeSTAR HS DNA polymerase(日本TAKARA公司,R010B)引物合成由上海捷瑞公司完成。
1.3 PCA、癌旁正常组织全基因外显子区突变位点筛查
1.3.1 PCA组织全基因外显子区SNPs检测 采用液相杂交捕获技术筛查PCA基因外显子区SNPs 突变位点。 组织提取及基因组DNA纯化 收集石蜡包埋的PCA、癌旁正常组织各50 g,按照Tiangen石蜡包埋组织DNA提取试剂盒说明书操作进行DNA的提取及纯化。琼脂糖凝聚电泳检测基因组DNA的完整性,采用Nanodrop 1000仪对基因组DNA的浓度进行定量,选择浓度>10 ng/μL的基因组DNA备用。采用Gnomegen DNA文库测序制备试剂盒进行文库构建。将样品基因组DNA进行随机打断,获得片段化DNA,断裂要求:300~500 bp片段,以400 bp为中心峰值;对片段化DNA进行末端修复和加dA尾,磁珠纯化后,在纯化DNA样品中加入连接接头,再次进行磁珠纯化,对接头纯化DNA样品进行PCR扩增,对PCR产物再次进行磁珠纯化,制成300~500 bp片段文库,-20 ℃保存备用。 探针杂交、捕获与洗脱 人工合成探针(SeqCap EZ Library Probes)由南京科维思生物科技有限公司完成。将扩增DNA文库样品与制备好的带生物素的自主定制探针加入至适当的液相杂交体系中,47 ℃、72 h温浴,完成杂交。然后用链霉亲和素磁珠对杂交产物进行序列捕获,将目标DNA(带生物素)绑定到磁珠上。之后用三种wash buffer对磁珠进行洗脱,洗脱后的目标DNA溶液-20 ℃保存。以洗脱后目标DNA溶液为模板,进行连接介导的PCR(LM-PCR)扩增,对捕获的目标基因进行富集。PCR产物经过Gnome size selector磁珠纯化后,-20 ℃保存。
1.3.2 外显子区SNPs突变位点比对 将捕获富集并验证的PCA、癌旁正常组织基因外显子区SNPs的DNA文库进行高通量测序,进行突变位点比对,筛选PCA基因外显子区突变位点。采用Illumina HiSeq 2000 平台测序,通过Illumina HiSeq 2000平台进行高通量测序。测序条件:读长为带标签的2×100 bp 双向测序,测序深度为>100×。
1.3.3 外显子区功能性突变位点生物信息学分析 采用fastqc软件对数据质量做质控,然后采用bwa软件与参考基因组hg19作比对,将比对结果手动去除重复后排序,再次采用GATK Indelrealigner对结果做重比对,接着采用GATK UnifiedGenotyper做变异检测,得到可信的SNP和indel。可信SNP和indel被用来做后续的annovar,在线SIFT(http://sift.jcvi.org/)和PolyPhe-2(http://genetics.bwh.harvard.edu/pph2/)注释,SIFT值小于0.05表示影响蛋白质功能。PolyPhe-2值越接近1,表示该SNP越能影响基因的表达。最后搜索OMIM数据库,查看样本疾病携带信息,并采用在线版HGMD数据库做了注释[12,13]。
1.3.4 外显子区功能性突变位点与临床易感性的关系 取保存在我院病理科的50例PCA患者(PSA值4.3~57.8 ng/mL,平均11.47 ng/mL;病理Gleason评分6~9分,平均7.28分)的PCA、癌旁正常组织,提取DNA,步骤同上。针对rs16941、rs45483396、rs121912655、rs7374710四个SNPs位点利用primer 6.0软件设计对应上下游引物(见表1)。引物由上海捷瑞公司合成。PCR扩增目的片段。反应体系50 μL,包括5×PS Buffer 10 μL,dNTP Mix (2.5 mmol/L each)4 μL,上下游引物(10 μmol/L)各1 μL,PrimeSTAR HS DNA polymerase 0.5 μL,DNA模板20 ng,加ddH2O至50 μL。反应条件:98 ℃、5 min,98 ℃、10 s,55 ℃ 、10 s,72 ℃、30 s,共30个循环,72 ℃、8 min,4 ℃保存。采用Sanger一代测序目的片段碱基并进行blast比对。

表1 扩增各SNPs位点目的片段引物序列及扩增长度
1.4 统计学方法 采用SAS9.1.3统计软件进行数据处理。组间各基因型频率的差异比较采用χ2检验,用比值比(OR)及95%可信区间(CI)表示各基因型的风险度,所有统计检验均为双侧概率检验。P<0.05为差异具有统计学意义。
2 结果
采用液相杂交捕获技术及高通量测序法比对PCA及癌旁正常组织,共检测到PCA外显子区17个SNPs突变位点,见表2。
采用PolyPhe-2及SIFT在线分析软件分析17个SNPs突变位点的生物学信息,共筛选到4个存在潜在差异功能SNPs,分别为BRCA1 rs16941、MET rs45483396、P53 rs121912655、COPB2 rs7374710。
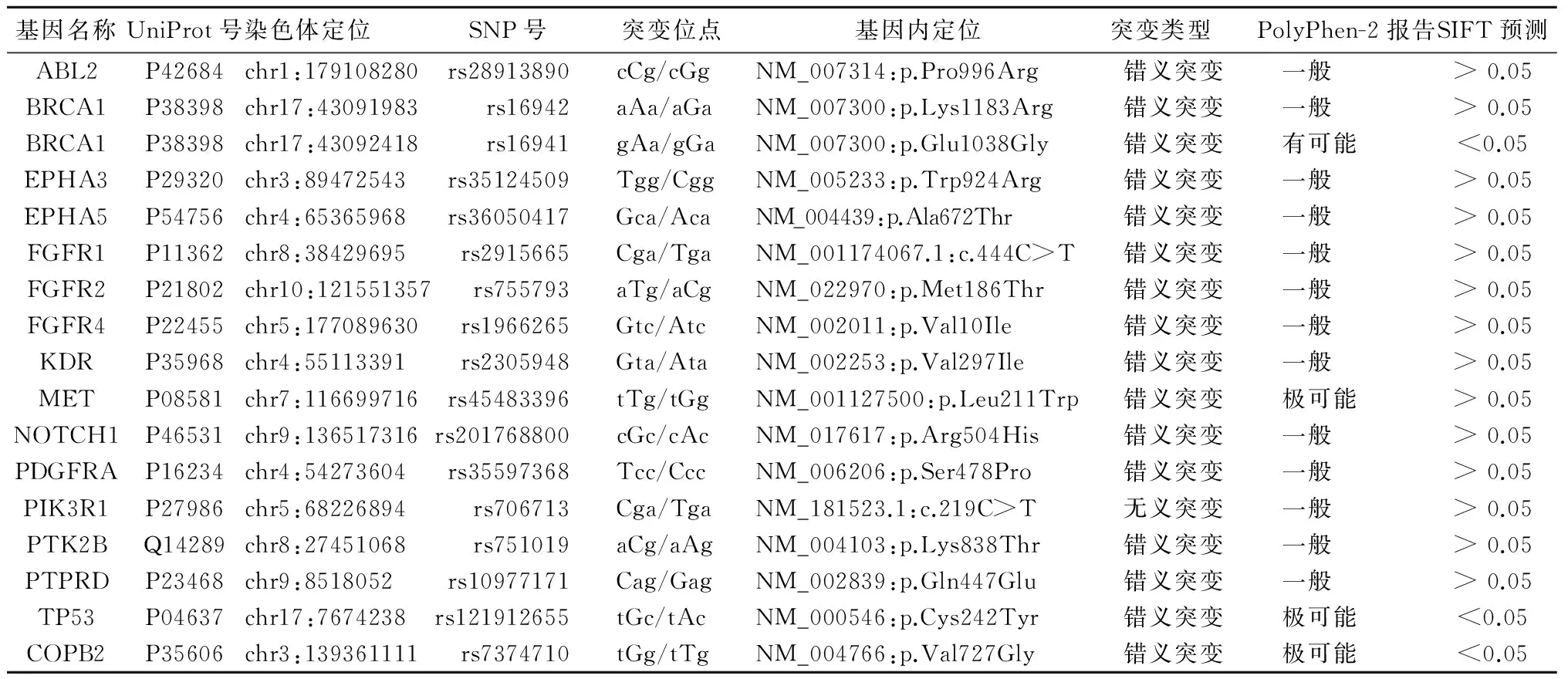
表2 PCA外显子区17个SNPs突变位点比较
50例PCA患者癌组织及癌旁正常组织分析结果显示,BRCA1 rs16941与COPB2 rs7374710两个SNP与PCA易感性相关(OR分别为 1.47, 1.27;95%CI分别为 1.14~1.72;1.20~1.41)。BRCA1 rs16941与COPB2 rs7374710 SNPs与PCA患者PSA值 及Gleason评分均呈正相关(OR分别为1.25,1.37;95%CI=1.10~1.84,1.20~2.34;P均< 0.01)。
3 讨论
基因研究在预测PCA易感性并揭示其发病机制方面一直扮演着重角色,而基因测序是最关键的环节。第一代测序技术以Sanger为代表,但其数据通量低、成本高,随着分子生物技术的发展及大数据时代的到来,其弊端日益显著。正是在这种背景下,下一代测序技术(NGS)应运而生,其高通量、低成本、时间短的优势特征已成为目前基因研究的主流[14]。
NGS在PCA研究中主要涉及检测基因突变、全基因组重测序、外显子组测序、表观遗传变异检测、RNA水平检测和染色体结构分析等[15]。液相杂交捕获技术可用于外显子测序,它是将感兴趣的基因组区域定制成特异性探针与基因组DNA进行杂交,将目标基因组外显子区域的DNA片段进行富集后再利用NGS技术进行测序的研究策略[16]。该技术能够针对某类疾病的所有基因变异情况进行非常深入的研究。
Spans等[17]运用NGS对激素敏感性PCA外显子区1 802个非同义SNPs和218个插入/缺失变异进行测序,证实PCA中雄激素受体(AR)和PTEN(两个基因存在突变,并建立了PCA细胞基因组蛋白质编码区的基因变异数据库。Robbins等[18]利用NGS对577个肿瘤相关基因的3 508个外显子进行靶向突变检测,结果发现8p22、10q23.31、13q13.1、13q14.11和13q14.12上存在局部纯合子缺失,定位于这些缺失区域的关键基因包括PTEN、BRCA2,C13ORF15和SIAH3。进一步发现在5p13.2p12、14q21.1、7q22.1和Xq12上有局部大量扩增,而定位于这些区域的关键基因包括AR、SKP2和FOXA1。Zuhlke等[19]用NGS对94例PCA样本进行外显子测序,发现1例高加索种族患者在NBN基因14号外显子存在2117C>G杂合子突变,该突变导致706密码子(S707X)翻译的提前终止,提示罕见的NBN突变与PCA易感性相关。Iacono等[20]选择60对局部或局部进展PCA样本进行NGS测序,结果鉴定出7种基因的8个遗传变异,分别是TP53 p.P72R(78%)、两个CSFR1 SNPs:rs2066934和rs066933(70%)、KDR p.Q472H(67%)、KIT p.M541L(28%)、PIK3CA p.I39M(19%)、MET p.V378I(10%)和FGFR3 p.F384L/p.F386L(8%)。并且证实TP53 p.P72R、CSFR1 SNPs、MET p.V378ISI四个SNPs与高级别PCA显著相关;FGFR3 p.F384L/p.F386L SNP与T2b以下PCA相关;MET p.V378I SNP与早期生化复发相关,提示这些SNPs与PCA发生、进展及复发关系密切,有望成为预测PCA病情进展的指标。
本研究对临床来源的PCA癌组织及癌旁组织进行测序,结果鉴定出16个基因的17个遗传变异,最后通过在线PolyPhe-2及SIFT分析筛选到4个潜在功能的SNPs(BRCA1 rs16941、MET rs45483396、P53 rs121912655、COPB2 rs7374710)。临床50例份PCA癌、癌旁组织标本中进行SNPs分型检测,结果发现BRCA1 rs16941、COPB2 rs7374710与PCA易感性有关。最后为了说明SNPs作为临床早期预测及后期监测的有效性,我们研究该SNPs与PCA常用评价指标PSA和Gleason评分之间的相关性,结果显示,BRCA1 rs16941,COPB2 rs7374710均与PSA值及Gleason评分呈正相关,揭示其可作为临床PCA早期诊断的分子标志物。本研究仍然存在一定的局限性,如前后纳入PCA样本量均较小、代表性不强,且组间存在异质性,可能存在假阳性或假阴性结果。
综上所述,PCA外显子区存在BRCA1 rs16941、COPB2 rs7374710功能性突变位点,携带有BRCA1 rs16941、COPB2 rs7374710突变位点的个体罹患PCA的可能性较高。
[1] Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016 [J]. CA Cancer J Clin, 2016,66(4):7-30.
[2] Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015 [J]. CA Cancer J Clin, 2016,66(2):115-132.
[3] Chen W, Zheng R, Zeng H, et al. Annual report on status of cancer in China, 2011 [J]. Chin J Cancer Res, 2015,27(1):2-12.
[4] Thompson IM, Pauler DK, Goodman PJ, et al. Prevalence of prostate cancer among men with a prostate-specific antigen level < or =4.0 ng per milliliter [J]. N Engl J Med, 2004,350(22):2239-2246.
[5] Gümü BH, Nee N, Gündüz MI, et al. Does asymptomatic inflammation increase PSA A histopathological study comparing benign andmalignant tissue biopsy specimens [J]. Int Urol Nephrol, 2004,36(4):549-553.
[6] Hochreiter WW. The issue of prostate cancer evaluation in men with elevated prostate-specific antigen and chronicprostatitis [J]. Andrologia, 2008,40(2):130-133.
[7] Thorpe A, Neal D. Benign prostatic hyperplasia [J]. Lancet, 2003,361(9366):1359-1367.
[8] Swanson GP, Thompson IM, Basler J. Current status of lymph node-positive prostate cancer: Incidence and predictors of outcome [J]. Cancer, 2006,107(3):439-450.
[9] 瞿旻,任善成,孙颖浩. PCA肿瘤标志物研究的新进展 [J]. 中华外科杂志,2015,53(4):317-320.
[10] Nordstrum T, Aly M, Eklund M, et al. A genetic score can identify men at high risk for prostate cancer among men with prostate-specific antigen of 1-3 ng/ml [J]. Eur Urol, 2014,65(6):1184-1190.
[11] Xu J, Mo Z, Ye D, et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4 [J]. Nat Genet, 2012,44(11):1231-1235.
[12] McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data [J]. Genome Res, 2010, 20(9):1297-1303.
[13] DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data [J]. Nat Genet, 2011, 43(5):491-498.
[14] Fullwood MJ, Wei CL, Liu ET, et al. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses [J]. Genome Res, 2009,19(4):521-532.
[15] 吴大鹏,康健. 下一代测序技术在PCA的研究进展 [J]. 现代泌尿外科杂志,2014,19(1):62-66.
[16] Holmes BJ, Westra WH. The expanding role of cytopathology in the diagnosis of HPV-related squamous cell carcinoma of the head and neck [J]. Diagn Cytopathol, 2014,42(1):85-93.
[17] Spans L, Atak ZK, Van Nieuwerburgh F, et al. Variations in the exome of the LNCaP prostate cancer cell line [J]. Prostate, 2012,72(12):1317-1327.
[18] Robbins CM, Tembe WA, Baker A, et al. Copy number and targeted mutational analysis reveals novel somatic events in metastatic prostate tumors [J]. Genome Res, 2011,21(1):47-55.
[19] Zuhlke KA, Johnson AM, Okoth LA, et al. Identification of a novel NBN truncating mutation in a family with hereditary prostate cancer [J]. Fam Cancer, 2012 ,11(4):595-600.
[20] Lo Iacono M, Buttigliero C, Monica V, et al. Retrospective study testing next generation sequencing of selected cancer-associated genes in resectedprostate cancer [J]. Oncotarget, 2016,7(12):14394-14404.
无锡市医院管理中心医学科研联合攻关项目( YGZXL1203;YGZXL 1318);无锡市科技发展资金(CSE31N1605)。
祝黎洁(E-mail: miniao1984@163.com)
10.3969/j.issn.1002-266X.2017.19.024
R737.25
B
1002-266X(2017)19-0077-04
2016-11-28)
