新型双- 1,4- 二氢吡啶的水相微波辅助合成
2017-05-17白少飞王进敏樊强文
白少飞, 王进敏, 樊强文, 闫 红*
(1. 北京工业大学 生命科学与生物工程学院,北京 100124; 2. 北京四环制药有限公司,北京 101113)
新型双- 1,4- 二氢吡啶的水相微波辅助合成
白少飞1, 王进敏2, 樊强文1, 闫 红1*
(1. 北京工业大学 生命科学与生物工程学院,北京 100124; 2. 北京四环制药有限公司,北京 101113)
采用微波辅助合成法,以四叔丁基溴化铵(TBAB)为催化剂,水为溶剂,3,3- 二乙氧基丙酸乙酯,胺和二醛(或醛和二胺)为原料,经Hantzsch反应合成了11个双- 1,4- 二氢吡啶化合物(I- 1~I- 7和II- 1~II- 4),其中I- 3~I- 7和II- 2~II- 4为新化合物,其结构经1H NMR,13C NMR和HR- MS(ESI)表征。以I- 1的合成为例,考察了相转移催化剂、微波功率、反应温度和反应时间对产率的影响。在最佳条件[TBAB 2.5 mol%,于100 W, 60 ℃微波反应30 min]下,I和II产率分别为70.9%~92.5%和79.8%~95.5%。
双- 1,4- 二氢吡啶; 微波辅助; 合成; 相转移催化; 条件优化
1,4- 二氢吡啶是一类含有平行双键的杂环己二烯类化合物,也是很多生物活性药物的基本结构单元,广泛存在于天然产物中。1,4- 二氢吡啶具有良好的生物药理活性,在医药、化工和生物等领域发挥了重要作用[1-4]。如1,4- 二氢吡啶类钙拮抗剂可使钙通道不被激活,阻碍钙离子进入细胞内,从而降低细胞内钙离子的含量,进而起到治疗高血压、心绞痛、动脉粥状硬化等血管性疾病的作用[5-6]。1,4- 二氢吡啶可作为原料合成吡啶类化合物,作为亚胺还原试剂,应用于还原氨化反应。此外,由于1,4- 二氢吡啶含有平行的不饱和双键,可在光照下发生光环合反应,生成笼型化合物[7-10]。
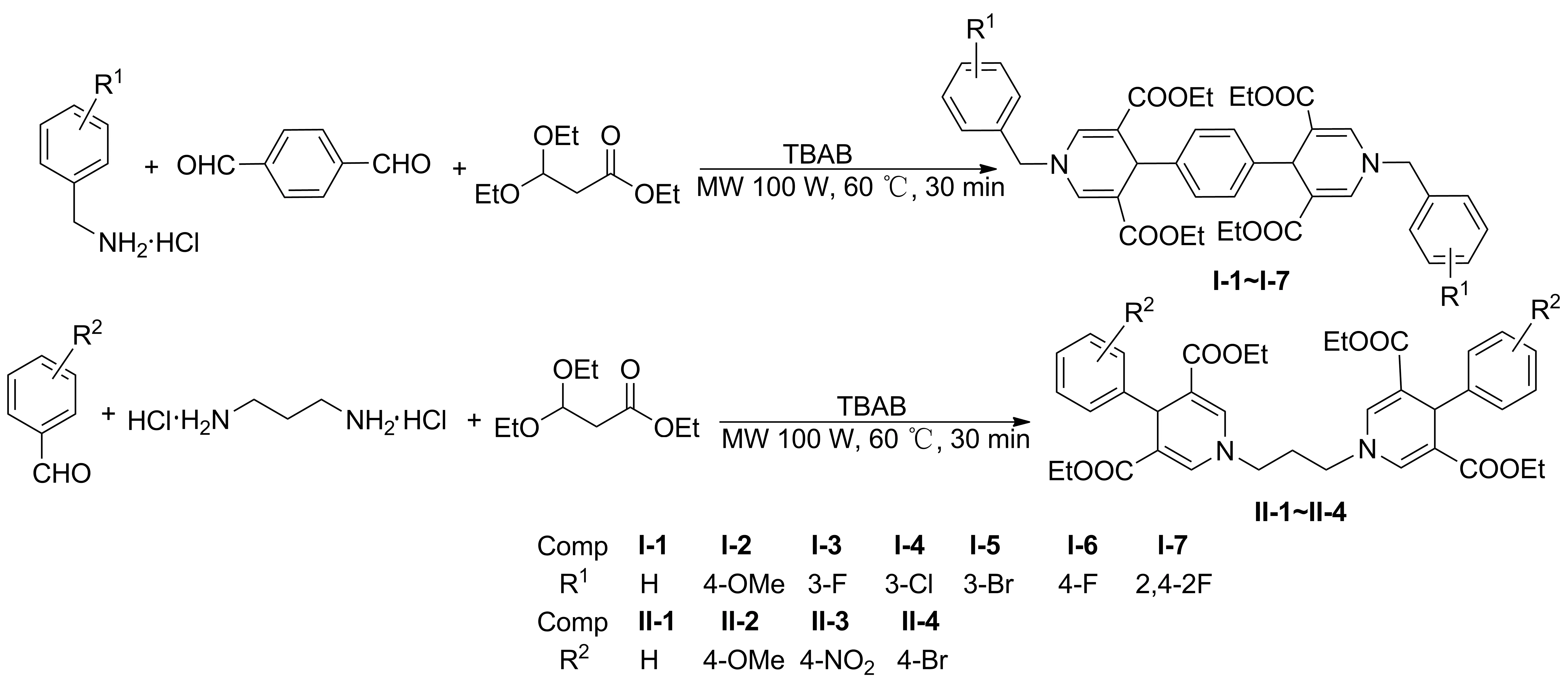
Scheme 1
双- 1,4- 二氢吡啶与1,4- 二氢吡啶具有结构相似性。由于连接两个二氢吡啶环的基团性质不同,可赋予1,4- 二氢吡啶类衍生物不同的生物药理活性。因此,对双- 1,4- 二氢吡啶的合成方法进行研究,有利于拓展1,4- 二氢吡啶类化合物在医药、化工和生物等领域的应用范围。
双- 1,4- 二氢吡啶的合成反应机理与1,4- 二氢吡啶的Hantzsch合成反应机理大致相同[11],均为醛和胺与β- 二羰基化合物发生亲核加成- 消除反应生成中间体;中间体再经过迈克尔加成- 消除反应和分子内环化反应得到最终产物。不同点在于:合成双- 1,4- 二氢吡啶时使用的原料为双官能团醛或胺[12-15],如对苯二甲醛、间苯二甲醛和1,3- 丙二胺等。Sueki等[16]以1,3- 丙二胺盐酸盐或苄胺盐酸盐为氮源,将对苯二甲醛和3,3- 二乙氧基丙酸乙酯溶于DMSO中,于90 ℃反应6~17 h,合成了双1,4- 二氢吡啶。该合成方法存在使用有机溶剂,反应时间较长,产率偏低,产物分离纯化过程繁琐等缺点。
水作为有机反应介质,具有无毒、环保和价廉易得等优点。以水作反应介质,还能通过调节反应体系的pH或添加相转移催化剂等方式以促进反应[17]。此外,由于有机产物大多在水中的溶解度较低,能从较大程度上降低产物损失,提高反应产率。
微波辅助反应方式已经被广泛应用于化工、医药和环保等领域[18-20]。微波促进有机合成反应具有低能耗、快速高效和产率高等优点。使用微波合成法优化双- 1,4- 二氢吡啶类衍生物的合成具有较大的研究意义。
鉴于此,本文采用微波辅助合成法,以四叔丁基溴化铵(TBAB)为催化剂,水为溶剂,3,3- 二乙氧基丙酸乙酯,胺和二醛(或醛和二胺)为原料,经Hantzsch反应合成了11个双- 1,4- 二氢吡啶化合物(I- 1~I- 7和II- 1~II- 4, Scheme 1),其中I- 3~I- 7和II- 2~II- 4为新化合物,其结构经1H NMR,13C NMR和HR- MS(ESI)表征。以I- 1的合成为例,考察了相转移催化剂、微波功率、反应温度和反应时间对产率的影响。
1 实验部分
1.1 仪器与试剂
X- 5型显微熔点仪;Bruker ARX 400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);Agilent G3250AA LC/MSD TOF system型高分辨质谱仪;Discover型微波合成仪。
所用试剂均为分析纯。
1.2 I和II的合成(以I- 1为例)
在单口烧瓶中加入苄胺盐酸盐2.86 g(20 mmol),蒸馏水50 mL,对苯二甲醛1.34 g(10 mmol), 3,3- 二乙氧基丙酸乙酯9.5 g(50 mmol)和TBAB 25 mol%,搅拌使其混合均匀;于100 W, 60 ℃微波反应30 min。用CH2Cl2萃取,有机相用少量H2O洗涤,用无水Na2SO4干燥,过滤,滤饼用乙酸乙酯和石油醚重结晶得淡黄色固体1,4- 双- (1- 苄基- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯)苯(I- 1)5.68 g,产率80.8%, m.p.165.0~166.3 ℃;1H NMRδ: 1.14(t,J=7.0 Hz, 12H), 4.02~4.10(m, 8H), 4.57(s, 4H), 4.85(s, 2H), 7.13(s, 4H), 7.25~7.42(m, 14H)。
用类似的方法合成I- 2~I- 7和II- 1~II- 4。
1,4- 双[1- (4- 甲氧基- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 2): 黄色固体,产率70.9%, m.p.168.2~169.9 ℃;1H NMRδ: 1.12(t,J=7.01 Hz, 12H), 3.84(s, 6H), 3.99~4.10(m, 8H), 4.50(s, 4H), 4.83(s, 2H), 6.93~6.95(d,J=8.40 Hz, 4H), 7.11(s, 4H), 7.20~7.28(q,J=7.72 Hz, 8H)。
1,4- 双[1- (3- 氟- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 3): 黄色固体,产率84.5%, m.p.170.5~172.8 ℃;1H NMRδ: 1.20(t,J=7.02 Hz, 12H), 4.04~4.15(m, 8H), 4.60(s, 4H), 4.94(s, 2H), 6.84~6.88(t,J=8.02 Hz, 2H), 6.98~7.00(d,J=8.02 Hz, 4H), 7.04~7.23(m, 8H), 7.27~7.38(t,J=7.30 Hz, 2H);13C NMRδ: 14.2, 37.2, 57.7, 60.2, 108.9, 113.2, 114.2, 115.0, 115.3, 122.7, 123.8, 129.2,130.9, 137.6, 138.6, 148.8, 161.9, 164.4; HR- MS(ESI)m/z: Calcd for C42H42N2O8F2{[M+Na]+}793.290 9, found 793.291 8。
1,4- 双[1- (3- 氯- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 4): 黄色固体,产率83.6%, m.p.174.6~176.8 ℃;1H NMRδ: 1.15(s, 12H), 3.98~4.15(m, 8H), 4.54(s, 4H), 4.85(s, 2H), 7.15(s, 4H), 7.22(s, 2H), 7.27(s, 4H), 7.33(s, 2H), 7.34(s, 4H);13C NMRδ: 14.1, 36.8, 57.5, 60.0, 109.5, 125.2, 127.3, 127.9, 128.5, 130.4, 135.0, 137.2, 138.3, 144.4, 166.9; HR- MS(ESI)m/z: Calcd for C42H42N2O8Cl2{[M+H]+}773.231 8, found 773.235 5。
1,4- 双[1- (3- 溴- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 5): 黄色固体,产率82.8%, m.p.175.5~177.8 ℃;1H NMRδ: 1.14(s, 12H), 3.98~4.14(m, 8H), 4.56(s, 4H), 4.85(s, 2H), 6.98~7.00(d,J=7.61 Hz, 2H), 7.05~7.07(d,J=7.61 Hz, 4H), 7.14(s, 4H), 7.22(s, 4H), 7.35~7.41(d,J=7.20 Hz, 2H);13C NMRδ: 14.1, 36.8, 57.6, 60.0, 109.5, 114.0, 114.2, 115.2, 115.4, 122.7, 127.9, 130.7, 130.8, 137.3, 138.8, 144.4, 161.9, 164.4, 166.9; HR- MS(ESI)m/z: Calcd for C42H42N2O8Br2{[M+H]+}861.231 8, found 861.232 0。
1,4- 双[1- (4- 氟- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 6): 黄色固体,产率87.2%, m.p.178.3~179.8 ℃;1H NMRδ: 1.14(t,J=7.21 Hz), 4.00~4.10(m, 8H), 4.57(s, 4H), 4.85(s, 2H), 6.98~7.00(d,J=8.80 Hz, 2H), 7.06~7.07(s, 4H), 7.14(s, 4H), 7.22(s, 4H), 7.36~7.41(q,J=7.21 Hz, 2H);13C NMRδ: 14.1, 36.8, 57.6, 60.0, 109.5, 114.0, 114.2, 115.2, 115.4, 122.7, 130.8, 137.2, 138.8, 144.4, 161.9, 164.4, 166.9; HR- MS(ESI)m/z: Calcd for C42H42N2O8F2{[M+Na]+}793.290 9, found 793.293 0。
1,4- 双[1- (2,4- 二氟- 苄基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]苯(I- 7): 黄色固体,产率92.5%, m.p.190.6~192.8 ℃;1H NMRδ: 1.14(t,J=7.00 Hz, 12H), 3.99~4.12(m, 8H), 4.57(s, 4H), 4.81(s, 2H), 6.88~6.94(m, 4H), 7.06(s, 4H), 7.14~7.23(m, 6H);13C NMRδ: 14.1, 36.8, 51.7, 60.0, 104.5, 109.5, 111.8, 11.20, 114.2, 115.2, 122.7, 127.7, 130.3, 137.0, 138.8, 144.4, 159.5, 161.7, 162.2, 164.3, 166.8; HR- MS(ESI)m/z: Calcd for C42H40N2O8F4{[M+Na]+}799.272 1, found 799.274 9。
1,3- 双- (4- 苯基- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯)丙烷(II- 1): 淡黄色固体,产率82.2%, m.p.169.0~172.3 ℃;1H NMRδ: 1.19(t,J=7.00 Hz, 12H), 2.13~2.17(t,J=7.21 Hz, 2H), 3.48~3.50(t,J=4.01 Hz, 4H), 4.04~1.14(m, 8H), 4.92(s, 2H), 7.04~7.30(m, 14H)。
1,3- 双- [4- (4- 甲氧苯基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]丙烷(II- 2): 黄色固体,产率79.8%, m.p.175.9~177.8 ℃;1H NMRδ: 1.20(t,J=7.01 Hz, 12H), 2.12~2.15(t,J=6.02 Hz), 3.48~3.52(t,J=8.01 Hz, 4H), 3.76(s, 6H), 4.07~1.14(m, 8H), 4.86(s, 2H), 6.81~6.83(d,J=8.01 Hz, 4H), 7.18(s, 4H), 7.20~7.22(d,J=8.01 Hz, 4H);13C NMRδ: 14.2, 30.9, 36.4, 51.8, 55.1, 60.1, 109.6, 113.4, 129.1, 136.5, 138.9, 158.2, 166.8; HR- MS(ESI)m/zCalcd for C39H46N2O10{[M-H]+}703.315 2, found 703.307 3。
1,3- 双- [4- (4- 硝基苯基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]丙烷(II- 3): 黄色固体,产率95.5%, m.p.173.8~175.3 ℃;1H NMRδ: 1.15(t,J=7.20 Hz, 12H), 2.22~2.25(t,J=6.61 Hz), 3.62~3.66(t,J=6.61 Hz, 4H), 4.02~4.11(m, 8H), 5.03(s, 2H), 7.28(s, 4H), 7.44~7.46(d,J=8.00 Hz, 2H), 7.72~7.73(d,J=7.61 Hz, 2H), 8.02~8.04(d,J=8.00 Hz, 2H), 8.13(s, 2H);13C NMRδ: 14.1, 30.6, 37.5, 51.8, 60.4, 108.6, 121.7, 123.0, 128.8, 134.6, 137.5, 148.4, 148.6, 166.2; HR- MS(ESI)m/z: Calcd for C37H40N4O12{[M-H]+}731.264 3, found 731.258 6。
1,3- 双- [4- (4- 溴苯基)- 1,4- 二氢吡啶- 3,5- 二羧酸乙酯]丙烷(II- 4): 黄色固体,产率85.8%, m.p.170.5~172.8 ℃;1H NMRδ: 1.19(t,J=7.21 Hz, 12H), 2.11~2.15(t,J=6.80 Hz), 3.48~3.52(t,J=7.01 Hz, 4H), 4.08~1.12(m, 8H), 4.89(s, 2H), 7.17~7.19(d,J=8.02 Hz, 8H), 7.40(s, 4H);13C NMRδ: 14.2, 30.7, 36.3, 51.5, 55.6, 60.1, 109.5, 110.1, 113.4, 129.1, 136.5, 137.3, 158.2, 166.8, 166.9; HR- MS(ESI)m/zCalcd for C37H40N2O8Br2{[M-H]+}798.115 1, found 798.116 6。
2 结果与讨论
2.1 合成反应条件优化
以I- 1的合成为例,考察了相转移催化剂,微波功率,反应时间和反应温度对产率的影响。
(1) 相转移催化剂
表1为相转移催化剂对I- 1产率的影响。由表1可见,相转移催化剂对I- 1产率影响较为显著。不使用相转移催化剂,反应难以进行;以环糊精和冠醚作相转移催化剂,催化效果不明显;季铵盐类相转移催化剂对反应的催化效果较好,TBAB和苄基三甲基溴化铵(BAB)催化下,产率分别为80.8%和78.5%,远高于文献产率(48.0%[16])。因此,选择TBAB为相转移催化剂。

表1 催化剂种类对I- 1产率的影响aTable 1 The effect of catalysts on the yield of I- 1
a反应条件同1.2。
此外,我们还研究了TBAB用量对I- 1产率的影响,结果见表2。由表2可见,当催化剂用量为2.5 mol%时,产率80.8%,用量超过2.5 mol%,产率基本维持不变。因此,TBAB的最佳用量为2.5 mol%。

表2 催化剂用量对I- 1产率的影响aTable 2 The amount of catalyst on the yield of I- 1
aTBAB为催化剂,其余反应条件同表1。
(2) 微波功率和反应温度
表3为微波功率和反应温度对I- 1产率的影响。由表3可见,当反应温度为30 ℃时,产率较低(50%左右);温度升高,产率显著提高,最高产率达75.6%。由表3还可见,提高反应功率,产率也随之上升,当微波功率为20 W时,产率小于50%;当微波功率为100 W时,最低收率也大于60%。因此,最佳反应温度为60 ℃,微波功率为100 W。

表3 微波功率和反应温度对I- 1产率的影响aTable 3 The effects of reaction temperature and microwave power on the yield of I- 1
a微波反应时间为10 min,其余反应条件同表2。
(3) 反应时间
表4为反应时间对I- 1产率的影响。由表4可见,延长反应时间,产率提高;当反应时间为30 min时,产率80.8%;继续延长反应时间,产率趋于稳定。因此,最佳反应时间为30 min。

表4 反应时间对I- 1产率的影响aTable 4 The effect of reaction time on the yield of I- 1
a反应温度为60 ℃,微波功率为100 W,其余反应条件同表3。
综上所述,I- 1的最佳合成条件为:TBAB 2.5 mol%,于100 W, 60 ℃微波反应30 min, I- 1产率80.8%。
2.2 最佳合成条件的反应拓展
(1) I的合成
表5为最佳合成条件在I- 2~I- 7的合成中的拓展结果。由表5可见,本文所用的合成方法的产率远高于文献值[16],且只需经萃取、浓缩和重结晶后即可得到终产物。由表5还可见,I的产率与苄胺上取代基的类型与位置有直接关系。当R为供电子基时,I的产率较低,如I- 1(R=H)产率为80.8%, I- 2(R=4- OMe)产率为70.9%;当R为吸电子基时,I的产率较高。吸电子基团的吸电子能力越强,产率越高。吸电子基团位于苯环的2- , 4- 位时,对产率的影响大于3- 位。如I- 6(R=4- F)产率为87.2%, I- 3(R=3- F)产率为84.5%。

表5 最佳合成条件在I- 2~I- 7合成反应中的拓展Table 5 Development of optimized conditions on the synthesis of I- 2~I- 7
(2) II的合成
表6为将最佳合成条件在II- 2~II- 7的合成中的拓展结果。由表6可见,用本文方法合成II- 1,产率高达82.2%,远高于文献值(49%)[16]。后处理时仅涉及萃取、浓缩、重结晶等简单步骤即可得到产物II,不需要使用柱层析纯化。由表6还可见,II的产率与芳香醛上取代基的电负性有直接关系。当R为供电子基团时,II产率降低。如II- 1(R=H)产率为82.2%, II- 2产率降至79.8%。当R为吸电子基团时,II产率随着基团吸电子能力的增强而提高。当R为吸电子能力较弱的Br时,II产率提高了约4%;当R为吸电子能力较强的NO2时,II产率提高13%以上。这可能是因为处于芳香醛对位的吸电子基团能够通过诱导效应和共轭效应降低醛基碳的电子云密度,增大碳原子的正电性,有利于亲核反应的进行;而对位取代的供电子基团通过诱导效应和共轭效应使碳原子的电子云密度增大,不利于亲核取代反应的进行,从而导致产率下降。
表6 最佳合成条件在II- 1~II- 4合成反应中的拓展Table 6 Development of optimized conditions on the synthesis of II- 1~II- 4
采用微波辅助合成法,以四叔丁基溴化铵(TBAB)为催化剂,水为溶剂,3,3- 二乙氧基丙酸乙酯,胺和二醛(或醛和二胺)为原料,经Hantzsch反应合成了11个双- 1,4- 二氢吡啶化合物(I- 1~I- 7和II- 1~II- 4, 其中I- 3~I- 7和II- 2~II- 4为新化合物)。在最佳条件[TBAB 2.5 mol%,于100 W, 60 ℃微波反应30 min]下,I和II产率分别为70.9%~92.5%和79.8%~95.5%。该方法具有绿色环保,后处理简单,产率较高等优点。
[1] Kappe C O. Biologically active dihydropyrimidones of the Biginelli- type- a literature survey[J].European journal of medicinal chemistry,2000,35(12):1043-1052.
[2] Gordeev M F, Patel D V, Gordon E M. Approaches to combinatorial synthesis of heterocycles:A solid- phase synthesis of 1,4- dihydropyridines[J].The Journal of Organic Chemistry,1996,61(3):924-928.
[3] Mannhold R, Jablonka B, Voigt W,etal. Calcium- and calmodulin- antagonism of elnadipine derivatives:Comparative SAR[J].European journal of medicinal chemistry,1992,27(3):229-235.
[4] Mager P, Coburn R, Solo A,etal. QSAR,diagnostic statistics and molecular modelling of 1,4- dihydropyridine calcium antagonists:A difficult road ahead[J].Drug design and discovery,1992,8(4):273-289.
[5] Rafieepour S, Saghaie L, Fassihi A. Conformationalproperties of novel 1,2,3,4- tetrahydro- pyrimidinone (thione) derivatives:A DFT study[J].Journal of Reports in Pharmaceutical Sciences(J Rep Pharm Sci),2013,1(2):118-126.
[6] Peri R, Padmanabhan S, Rutledge A,etal. Permanently charged chiral 1,4- dihydropyridines:Molecular probes of L- type calcium channels:Synthesis and pharmacological characterization of methyl (ω- trimethylalkylammonium) 1,4- dihydro- 2,6- dimethyl- 4- (3- nitrophenyl)- 3,5- pyridinedicarboxylate iodide,calcium channel antagonists[J].Journal of medicinal chemistry,2000,43(15):2906-2914.
[7] Kempson J, Spergel S H, Guo J,etal. Novel tricyclic inhibitors of IκB kinase[J].Journal of medicinal chemistry,2009,52(7):1994-2005.
[8] Itoh T, Nagata K, Miyazaki M,etal. A selective reductive amination of aldehydes by the use of Hantzsch dihydropyridines as reductant[J].Tetrahedron,2004,60(31):6649-6655.
[9] Zhu X, Li W, Yan H,etal. Triplet phenacylimidazoliums- catalyzed photocycloaddition of 1,4- dihydropyridines:An experimental and theoretical study[J].Journal of Photochemistry and Photobiology A:Chemistry,2012,241:13-20.
[10] 朱晓鹤,倪成良,宋秀庆,等. 3,9- 二氮杂四星烷类化合物的合成研究[J].有机化学,2010,30(2):276-281.
[11] Hantzsch A. Ueber die synthese pyridinartiger verbindungen aus acetessigäther und aldehydammoniak[J].Justus Liebigs Annalen der Chemie,1882,215(1):1-82.
[12] Mukherjee A, Akhtar M S, Sharma V L,etal. Syntheses and bioevaluation of substituted dihydropyridines for pregnancy- interceptive activity in hamsters[J].Journal of medicinal chemistry,1989,32(10):2297-2300.
[13] Ghorbani C A, Zolfigol M A, Hajjami M,etal. Nano aluminium nitride as a solid source of ammonia for the preparation of Hantzsch 1,4- dihydropyridines and bis- (1,4- dihydropyridines) in waterviaone pot multicomponent reaction[J].Journal of the Brazilian Chemical Society,2011,22(3):525-531.
[14] Salehi H, Guo Q X. Synthesis ofsubstituted 1,4- dihydropyridines in water using phase- transfer catalyst under microwave irradiation[J].Synthetic communications,2004,34(23):4349-4357.
[15] Abdel L F, Mashaly M, Mekheimer R,etal. Heterocycle syntheses through ternary condensation of terephthalaldehyde with malononitrile and some nucleophiles[J].Zeitschrift für Naturforschung B,1993,48(6):817-820.
[16] Sueki S, Takei R, Zaitsu Y,etal. Synthesis of 1,4- dihydropyridines and their fluorescence properties[J].European Journal of Organic Chemistry,2014,(24):5281-5301.
[17] 何艳,齐红. 相转移催化反应[J].长春师范学院学报,2005,(7):37-38.
[18] Khadilkar B M, Gaikar V G, Chitnavis A A. Aqueous hydrotrope solution as a safer medium for microwave enhanced hantzsch dihydropyridine ester synthesis[J].Tetrahedron Letters,1995,36(44):8083-8086.
[19] 孙纳新. 微波技术及其应用[J].医药化工,2006,(4):44-50.
[20] Tu S, Miao C, Gao Y,etal. A novel cascade reaction of aryl aldoxime with dimedone under microwave irradiation:The synthesis ofN- hydroxylacridine[J].Synlett,2004,(2):255-258.
Synthesis of Novel Bis- 1,4- dihydropyridines Assisted by Microwave in Aqueous Phase
BAI Shao- fei1, WANG Jin- min2, FAN Qiang- wen1, YAN Hong1*
(1. College of Life Science and Biotechnology, Beijing University of Technology, Beijing 100124, China; 2.Sihuan Pharmaceutical Co., Ltd., Beijing 101113, China)
Eleven bis- 1,4- dihydropyridines(I- 1~I- 7 and II- 1~II- 4) were synthesized by Hantzsch reaction under the assistance of microwave in aqueous phase, using TBAB as the catalyst, 3,3- ethoxypropionate, amine and dialdehyde(or aldehyde and diamine) as the raw materials. Among them, I- 3~I- 7 and II- 2~II- 4 were novel compounds. The structures were characterized by1H NMR,13C NMR and HR- MS(ESI). The effects of the phase transfer catalyst, microwave power, temperature and time on the yield were investigated, using I- 1 as the example. The yields of I and II were 70.9%~92.5% and 79.8%~95.5%, respectively, under the optimized reaction condition[TBAB 2.5 mol%, irradiation at 100 W and 60 ℃ for 30 min].
bis- 1,4- dihydropyridine; microwave assistant; synthesis; phase transfer catalysis; condition optimization
2017- 01- 08;
2017- 04- 28
北京市自然科学基金重点项目(KZ201510005007)
白少飞(1990-),男,汉族,河北秦皇岛人,硕士研究生,主要从事有机合成的研究。 E- mail: 1164092499@qq.com
闫红,教授, E- mail: hongyan@bjut.edu.cn
O623.6; O623.7
A
10.15952/j.cnki.cjsc.1005- 1511.2017.05.17003
