LC-MS/MS 法同时检测生物检材中钩吻素子、钩吻素甲和钩吻素己
2017-05-13姬圣洁刘伟
姬圣洁,刘伟
(1.复旦大学基础医学院法医学系,上海 200032;2.司法部司法鉴定科学技术研究所上海市法医学重点实验室上海市司法鉴定专业技术服务平台,上海 200063)
·论著·
LC-MS/MS 法同时检测生物检材中钩吻素子、钩吻素甲和钩吻素己
姬圣洁1,2,刘伟2
(1.复旦大学基础医学院法医学系,上海 200032;2.司法部司法鉴定科学技术研究所上海市法医学重点实验室上海市司法鉴定专业技术服务平台,上海 200063)
目的建立准确、灵敏的生物检材中钩吻素子、钩吻素甲及钩吻素己的液相色谱-串联质谱(LCMS/MS)检测方法,并进行方法学验证。方法以士的宁为内标,血液、尿液及肝组织样品经1%氢氧化钠溶液碱化后用乙酸乙酯提取,采用ZORBAX SB-C18柱(150mm×2.1mm,5μm)分离,以甲醇-20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)为流动相进行梯度洗脱。定性定量分析采用电喷雾正离子化(ESI+)、多反应监测模式。结果血液、尿液及肝组织中钩吻素子、钩吻素甲和钩吻素己在相应的线性范围内线性良好,相关系数(r)>0.9950,检出限分别为0.1 ng/mL(或0.1 ng/g)、0.1ng/mL(或0.1 ng/g)及0.01ng/mL(或0.01ng/g),各生物碱提取回收率为61.9%~114.6%,准确度为92.4%~114.3%,日内、日间精密度的相对标准偏差均不超过11.0%。结论本方法选择性好、灵敏度高,适用于同时检测生物体液和组织中的钩吻素子、钩吻素甲和钩吻素己,可为钩吻中毒的临床诊治和法医学鉴定提供有效的技术支撑。
法医毒理学;液相色谱-串联质谱法;钩吻素子;钩吻素甲;钩吻素己;生物检材
钩吻(Gelsemium elegans Benth.)为马钱科胡蔓藤属植物胡蔓藤的全株,又名胡蔓藤、断肠草、大茶药、野葛、毒根等,主要分布在东南亚和我国的云南、贵州、福建、广东、广西、浙江、湖南等地。胡蔓藤属植物共有三种,除了钩吻外还有分布在北美地区的常绿钩吻(Gelsemium sempervirens Alt.)和沼泽茉莉(Gelsemium rankinii Small.)。在民间,钩吻多以外用为主,具有祛风、杀虫止痒、消肿拔毒的疗效,此外还有抗肿瘤、抗焦虑、消炎止痛、免疫调节、散瞳等功效[1-3]。钩吻为剧毒植物,临床上对人的消化系统、神经系统、循环和呼吸系统都有不同程度的损害,严重者可因呼吸麻痹而死亡[4]。吲哚类生物碱为胡蔓藤属植物的主要化学成分,也是其发挥药理作用和产生毒性的主要来源,目前在其中共发现121种吲哚类生物碱[5]。分布在亚洲地区的钩吻以钩吻素子(koumine)含量最高[6],钩吻素甲(gelsemine)次之,有研究[7,8]表明钩吻素己(gelsenicine)毒性更大(三者的化学结构见图1),分布在不同地区的钩吻化学成分会有些许差异。由于钩吻治疗量和中毒量非常接近,因使用不当、自杀、恶意投毒或治疗过程中发生中毒甚至死亡的案件时有发生[9],因此,有必要建立灵敏、可靠的分析方法检测生物检材中的钩吻生物碱。
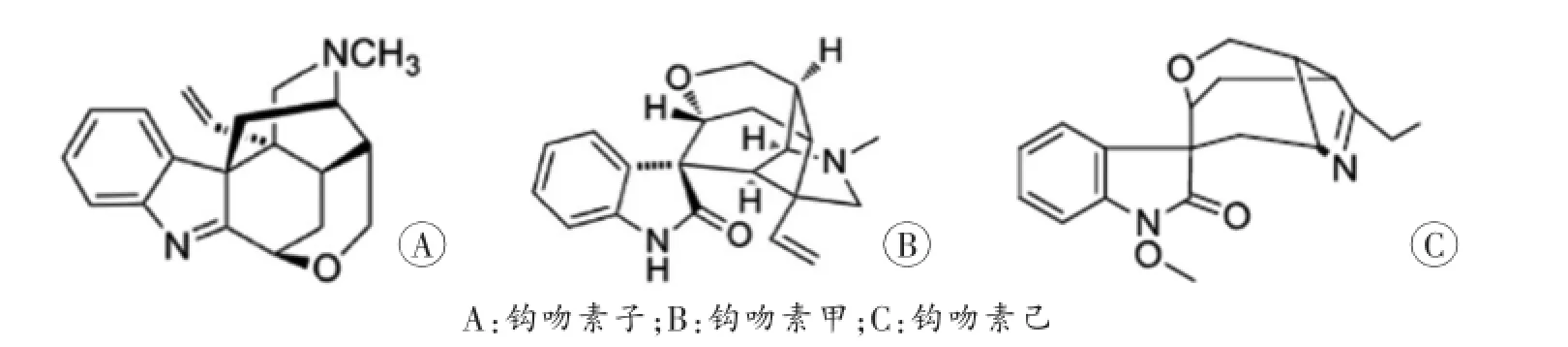
图1 钩吻素子、钩吻素甲和钩吻素己的化学结构
目前,生物检材中钩吻生物碱的分析方法主要有液相色谱(LC)法[10]、气相色谱-质谱联用(GC-MS)法[11,12]、气相色谱-串联质谱(GC-MS/MS)法[13]、液相色谱-串联质谱(LC-MS/MS)法[14-17]。与前几种方法相比,LC-MS/MS法将液相色谱的分离能力和质谱提供化合物结构信息的功能结合在一起,具有选择性强、灵敏度高的优势。现有文献中所报道的分析方法一般只检测钩吻素甲或(和)钩吻素子,适用的检材单一,难以满足钩吻中毒鉴定的需求。因此本研究拟建立同时检测多种生物检材(血液、尿液、肝组织)中钩吻素子、钩吻素甲和钩吻素己的LC-MS/MS法,旨在为钩吻中毒的临床诊治和法医学鉴定提供科学的技术支撑。
1 材料与方法
1.1 材料
1.1.1 仪器
AcquityTM超高效液相色谱仪(美国Waters公司),4000 Q TRAP四极杆-线性离子阱质谱仪(美国AB SCIEX公司),MD200-2氮气吹扫仪(杭州奥盛仪器有限公司),HR2860 Cucina搅拌机(荷兰皇家飞利浦电子公司),TDZ4-WS离心机(上海卢湘仪离心机仪器有限公司),Centrifuge 5810R冷冻离心机(德国Eppendorf公司),BSA124S电子天平[赛多利斯科学仪器(北京)有限公司],XW-80A旋涡混合器(上海医大仪器有限公司),Milli-Q超纯水系统(美国Millipore公司)。
1.1.2 试剂
对照品钩吻素子(纯度99.56%)、钩吻素甲(纯度98.36%)及钩吻素己(纯度99.53%)均购自成都曼思特生物科技有限公司,内标士的宁对照品(以97%计)购自中国药品生物制品检定所。
甲醇、乙腈、乙酸铵(纯度≥99.0%)、50%甲酸溶液,均为HPLC级,购自美国Fluka公司;氢氧化钠、乙酸乙酯、乙醚、氯仿,均为分析纯,购自上海凌峰化学试剂有限公司;硼砂购自优耐德引发剂(上海)有限公司;实验用水为超纯水,由Milli-Q超纯水系统制得。
空白血液(经肝素抗凝)、空白尿液均来源于健康志愿者(均已知情同意),空白肝为市售的新鲜猪肝。
1.1.3 溶液配制
对照品溶液:分别精密称取钩吻素子、钩吻素甲和钩吻素己对照品5mg,置于5mL容量瓶中,加入甲醇溶解并定容至刻度,配制成质量浓度均为1mg/mL的钩吻素子、钩吻素甲、钩吻素己甲醇储备液,密封后置于-20℃冰箱中冷冻保存,备用。实验中所需要的不同浓度的对照品溶液均从上述储备液中用甲醇稀释获得。
内标溶液:精密称取士的宁对照品10mg置于10mL容量瓶中,加入甲醇溶解并定量至刻度,配制成质量浓度为1mg/mL的士的宁甲醇储备液。取适量士的宁储备液,用甲醇稀释得2μg/mL的内标工作液。士的宁储备液和工作液密封后在4℃冰箱中保存,待用。
20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈):称取乙酸铵0.77 g置于500mL容量瓶中,加入适量超纯水溶解,再加入50%甲酸溶液0.92g和乙腈25mL,以超纯水定容至刻度,充分混匀即可。
复溶溶液:V(甲醇)∶V[20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)]=70∶30。
1.2 实验方法
本实验对色谱柱、流动相、质谱母离子和子离子、内标以及样品前处理中的萃取溶剂、碱化试剂、复溶溶液进行了优化,以下是优化后的实验条件。
1.2.1 样品前处理
取血液或尿液样品0.5mL,加入2μg/mL士的宁工作液10μL、1%氢氧化钠溶液50μL,涡旋混合1min(混合物pH值约为8.2),再加入乙酸乙酯3mL萃取,涡旋2min后,以离心半径12 cm,3 000 r/min,离心3min。取上清液于50℃下在氮气吹扫仪上吹干,加入复溶溶液100μL,混匀后供LC-MS/MS分析。
将生物组织剪碎并研磨成匀浆,称取组织匀浆0.5 g,加入2μg/mL士的宁工作液10μL、1%氢氧化钠溶液800μL,涡旋混合1min(混合物pH值约为9.0),再加入乙酸乙酯3mL萃取,涡旋3min后,以离心半径12 cm,3 000 r/min,离心3min。取上清液于50℃下在氮气吹扫仪上吹干,加入复溶溶液200μL,涡旋均匀后转移至1.5mL离心管中,于-20℃冰箱中放置30min,4℃下以离心半径9.5 cm,13 000 r/min,离心2min,取上清液供LC-MS/MS分析。
1.2.2 仪器条件
1.2.2.1 色谱条件
色谱柱为ZORBAX SB-C18柱(150mm×2.1mm,5μm),购自美国Agilent公司,柱温为室温,前接ZORBAX-Extend-C18保护柱(美国Agilent公司);流动相A为20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈),B为甲醇;流速为0.2mL/min;梯度洗脱程序为0~0.5min 10%B,0.5~0.6min 10%~40%B,0.6~1.5min 40%B,1.5~1.6min 40%~70%B,1.6~5.5min 70%B,5.5~5.6min 70%~10%B,5.6~8.0min 10%B;进样量为5μL。
1.2.2.2 质谱条件
电喷雾离子源采用正离子模式(ESI+),多反应监测(multiple reaction monitor,MRM),离子源电压5 500 V,离子源温度500℃,气帘气30 psi,雾化气40psi,辅助气50 psi。
在上述质谱条件下,用针泵分别将单独含有10 ng/mL钩吻素子、钩吻素甲、钩吻素己和士的宁的甲醇溶液,以10μL/min的流速注入质谱,优化质谱参数。其中化合物的两对母离子-子离子对用于定性分析,第一对离子对用于定量分析,LC-MS/MS参数见表1。
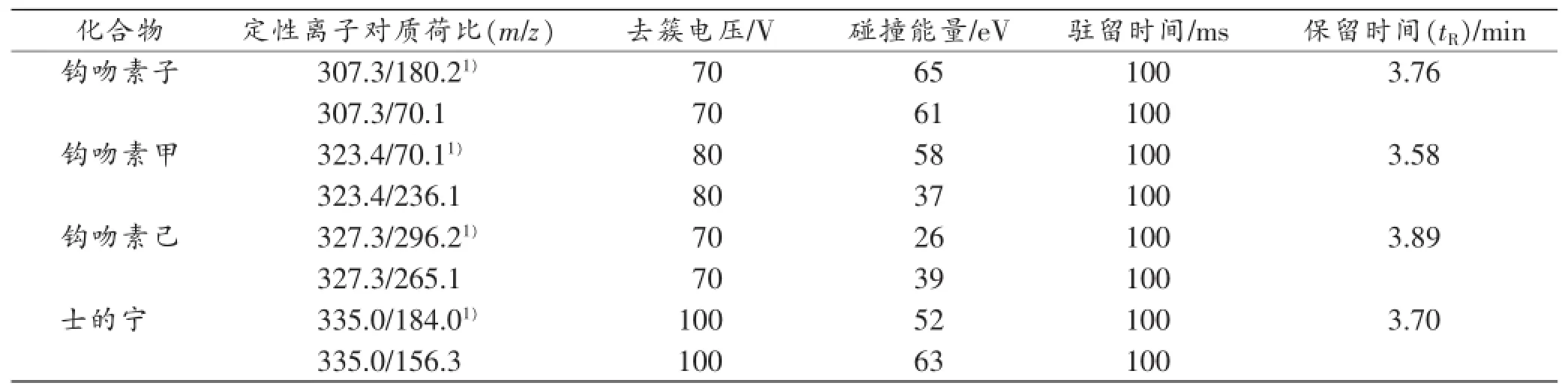
表1 钩吻素子、钩吻素甲、钩吻素己和内标士的宁的LC-MS/MS参数
1.3 方法学验证
对所建方法的选择性、线性、灵敏度、准确度、精密度、提取回收率、基质效应和稳定性方面进行验证。
选择性:取10份来源于不同个体的空白血液、尿液和肝组织,按照“1.2.1样品前处理”方法操作,在“1.2.2仪器条件”下进样分析;另取相同空白基质添加钩吻素子、钩吻素甲、钩吻素己和士的宁对照品,同法处理后供LC-MS/MS分析。要求空白基质中的内源性物质对待测物及内标没有干扰。
线性和灵敏度:分别取空白血液、尿液和肝组织0.5mL(g)若干份,添加不同浓度的钩吻素子、钩吻素甲和钩吻素己混合标准溶液以及内标对照品溶液配制成系列样品,每个浓度平行2份。样品按照“1.2.1样品前处理”方法操作,在“1.2.2仪器条件”下进样分析。以3种待测物含量(ng/mL或ng/g)为横坐标,待测物峰面积与内标峰面积之比为纵坐标作线性回归(权重系数1/x),得到回归方程和相关系数,并将信噪比S/N≥3和S/N≥10的浓度分别作为检出限(limit of detection,LOD)和定量限(limit of quantitation,LOQ)。
准确度和精密度:分别取空白血液、尿液和肝组织0.5mL(g)若干份,添加不同浓度的钩吻素子、钩吻素甲和钩吻素己混合标准溶液以及内标对照品溶液,配制成低、中、高浓度的质控样品,每个浓度平行6份。样品按照“1.2.1样品前处理”方法操作,在“1.2.2仪器条件”下进样分析。根据当日工作曲线计算待测物浓度,考察方法的准确度和日内精密度,同法测定4d,计算日间精密度。
提取回收率和基质效应:制备低、中、高浓度的质控样品,每个浓度平行6份,根据“1.2.1样品前处理”方法操作,在“1.2.2仪器条件”下进样分析,测得各待测物峰面积为A1;相同空白基质前处理后再添加待测物,所得峰面积为A2;用复溶溶液稀释标准品溶液至相同进样浓度测得峰面积为A3;提取回收率(%)= A1/A2×100%,基质效应(%)=A2/A3×100%。
稳定性:制备低、中、高浓度的质控样品,每个浓度平行6份,分别考察室温条件下放置24h、样品处理后于进样盘中放置24h、3次冻融循环(-20℃冰箱中放置21 h,转移到室温下放置3 h,反复3次)、于-20℃冰箱中冷冻保存1个月这4种条件下待测物的稳定性。测定浓度均值与理论浓度的相对偏差绝对值不超过20%,说明待测物的稳定性良好。
2 结果与讨论
2.1 方法的优化
2.1.1 仪器条件的选择
2.1.1.1 色谱柱的选择
本实验考察了4种色谱柱对钩吻素子、钩吻素甲、钩吻素己和内标的分离效果,发现使用Allure PFP Propyl柱(100mm×2.1mm,5μm)分离时,各待测物峰形过宽且出峰时间较晚;用Capcell Pak C18柱(250mm× 2.0mm,MGⅡ5μm)分离时,待测物的峰形出现严重拖尾;经Phenomenex Luna C18(2)柱(150mm×2mm,3μm)分离时,待测物峰形较宽、不对称;而最终选用的ZORBAX SB-C18柱(150mm×2.1mm,5μm)可使各待测物分离,峰形较好且灵敏度较高。
2.1.1.2 流动相的选择
流动相的组成直接影响分离效果、离子化效率和灵敏度。本研究考察了乙腈-0.1%甲酸、乙腈-20mmol/L乙酸铵缓冲溶液、乙腈-20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)、甲醇-0.1%甲酸、甲醇-20mmol/L乙酸铵缓冲溶液、甲醇-20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)共6种流动相体系,发现将乙腈作为有机相时待测物峰形很差,而甲醇-20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)梯度洗脱效果较佳,各待测物峰形较窄且出峰时间合适。
2.1.1.3 质谱条件的优化
分别取适量钩吻素子、钩吻素甲、钩吻素己以及内标士的宁储备液,用甲醇稀释成10ng/mL的对照品溶液,在ESI+模式下,经针泵以10μL/min的流速注入质谱。通过全扫描方式可得到钩吻素子、钩吻素甲、钩吻素己和士的宁的准分子离子峰作为其母离子,质荷比(m/z)分别为307.3、323.4、327.3、335.0。在母离子扫描的基础上进行子离子全扫描,选择至少两个丰度较高的子离子与其母离子组成母离子-子离子对,再优化去簇电压和碰撞能量。最终选择两对母离子-子离子对作为定性离子对,其中丰度较高的一对作为定量离子对。
2.1.1.4 内标的选择
内标法定量是通过测量待测物与内标峰面积的比值进行计算的,在一定程度上消除了仪器不稳定、进样量不完全相同等原因引起的误差。本研究考察了吴茱萸碱、士的宁及麦角新碱3种化合物作内标时的出峰情况,结果表明,在本研究的LC-MS/MS条件下吴茱萸碱在8min内不出峰,麦角新碱峰形较差且容易残留在液相系统里,而士的宁与被分析物极性相似、不发生化学反应,峰形良好,与待测物能够很好地分离且保留时间与各待测物接近,能够很好地起到校正的作用。
2.1.2 样品前处理方法的优化
2.1.2.1 萃取溶剂的选择
在LC-MS/MS分析中,生物样品前处理常用液液萃取法、蛋白沉淀法和固相萃取法,需要结合检材的特点和待测物的理化性质来选择。钩吻素子、钩吻素甲和钩吻素己难溶于水,可溶于氯仿、乙酸乙酯等有机溶剂,选择液液萃取法进行前处理。本实验比较了乙醚、乙酸乙酯和氯仿3个溶剂的提取效率,血液样品中氯仿>乙酸乙酯>乙醚,尿液样品中乙酸乙酯>氯仿>乙醚,但氯仿毒性较大,综合考虑选择乙酸乙酯作为提取溶剂,经方法验证表明此前处理方法能得到满意的灵敏度和提取回收率。
2.1.2.2 碱化试剂的选择
钩吻素子、钩吻素甲和钩吻素己都属于生物碱类,部分以生物碱盐的形式存在。若要提取完全,通常需要加入碱化试剂使生物碱变成游离状态。本实验比较了硼砂和氢氧化钠溶液的碱化效果,0.5mL血液或尿液样品中加入pH=9.2的硼砂缓冲溶液1mL后用乙酸乙酯萃取,可获得较为满意的提取回收率,但采用此法处理0.5 g肝组织样品时,提取回收率小于10%,碱化不完全导致提取效率太低。用1%氢氧化钠溶液50μL替代pH=9.2的硼砂缓冲溶液1mL加入血液或尿液(混合物pH值约为8.2),提取回收率为67.6%~97.2%,二者的碱化效果相差不大。0.5 g肝组织加入1%氢氧化钠溶液800μL(混合物pH值约为9.0),提取回收率可提高到61.9%~114.6%。选用的氢氧化钠溶液浓度较低,在生物检材中产生的固化作用不严重,对实验结果没有明显的不良影响,因此经过综合比较,选择1%氢氧化钠溶液作为碱化试剂。
2.1.2.3 复溶溶液的选择
样品复溶溶液的组成会影响待测物的色谱行为及离子化效率。本实验比较了4种不同比例的流动相溶液,即甲醇与20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)的体积比分别为100∶0、70∶30、40∶60、10∶90,作为复溶溶液时进样分析的出峰情况。实验结果表明,甲醇的比例越高,各组分的峰形更好、丰度更高。但肝组织中含有较多的脂溶性成分,甲醇相比第二种比例的流动相溶解了更多的脂溶性杂质,因此最终选择甲醇与20mmol/L乙酸铵缓冲溶液(含0.1%甲酸和5%乙腈)的体积比为70∶30时的流动相作为复溶溶液。
处理血液和尿液样品时复溶溶液体积为100μL,而处理肝组织样品时改为200μL,因为肝组织中脂溶性杂质较多,多次进样后可能会影响色谱柱的柱效导致峰形变差、灵敏度下降,在保证灵敏度的前提下增大复溶溶液的体积可减轻杂质的影响。
2.2 方法学验证
2.2.1 选择性
空白血液、尿液和肝组织的MRM色谱图见图2~4。以血液样品为例,空白血液中加入各待测物后含钩吻素子(10 ng/mL)、钩吻素甲(10 ng/mL)、钩吻素己(1 ng/mL)以及内标士的宁(40ng/mL),按照“1.2.1样品前处理”方法操作,在“1.2.2仪器条件”下进样分析,结果见图5,钩吻素子、钩吻素甲、钩吻素己和士的宁的保留时间分别为3.76、3.58、3.89、3.70min。比较两者的MRM色谱图可知空白样品在待测物及内标的出峰位置无响应,说明空白基质的内源性物质对分析物的测定没有干扰。
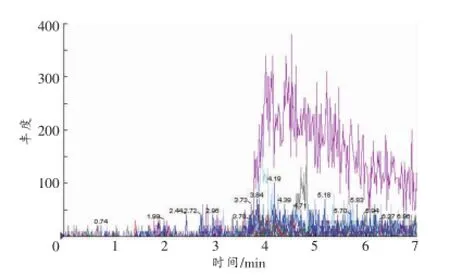
图2 空白血液的MRM色谱图
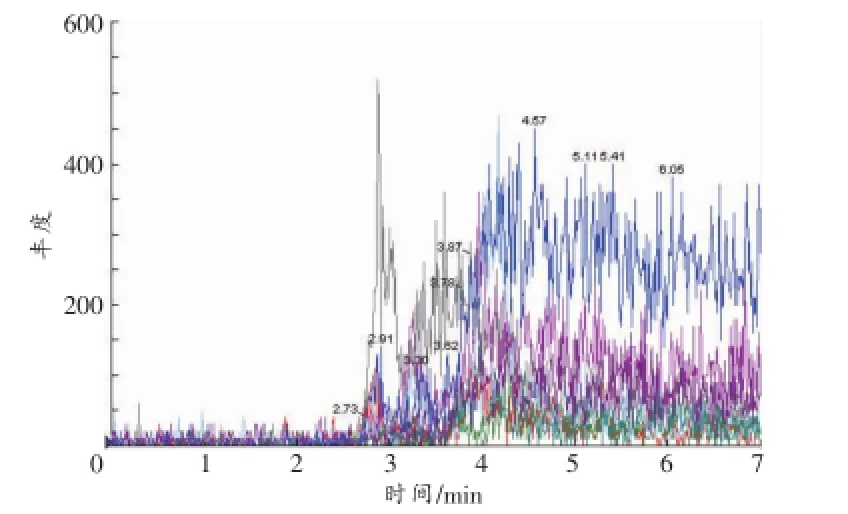
图3 空白尿液的MRM色谱图
图4 空白肝组织的MRM色谱图
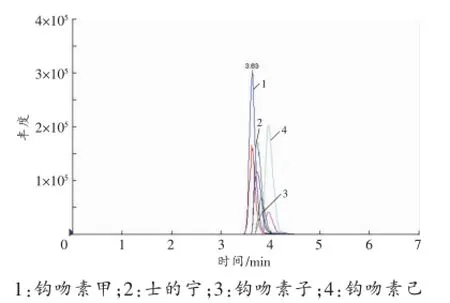
图5 空白血液中添加待测物后的MRM色谱图
2.2.2 线性范围、LOD和LOQ
血液、尿液及肝组织中钩吻素子、钩吻素甲和钩吻素己在相应的线性范围内线性良好,相关系数(r)>0.9950,LOD分别为0.1ng/mL(或0.1ng/g)、0.1ng/mL(或0.1ng/g)及0.01ng/mL(或0.01ng/g),结果见表2,本方法的灵敏度和线性范围能够满足临床诊治和法医毒物分析的需要。
2.2.3 准确度和精密度
对于生物样品分析,要求方法的准确度应在85%~115%,在LOQ附近可放宽至80%~120%;方法精密度的相对标准偏差(relative standard deviation,RSD)应控制在15%,在LOQ附近RSD应小于20%。本研究的结果见表3,各待测物的准确度在92.4%~114.3%,日内、日间精密度的RSD不超过11.0%,表明该方法的准确度和精密度均满足要求。
2.2.4 提取回收率与基质效应
本方法提取回收率和基质效应的实验结果见表3,各待测物的提取回收率为61.9%~114.6%,表明乙酸乙酯液液萃取的前处理方法可达到较满意的提取效率。基质效应表现为轻微抑制,可能与样品基质、流动相、样品前处理过程、色谱分离效果和离子化等因素有关。
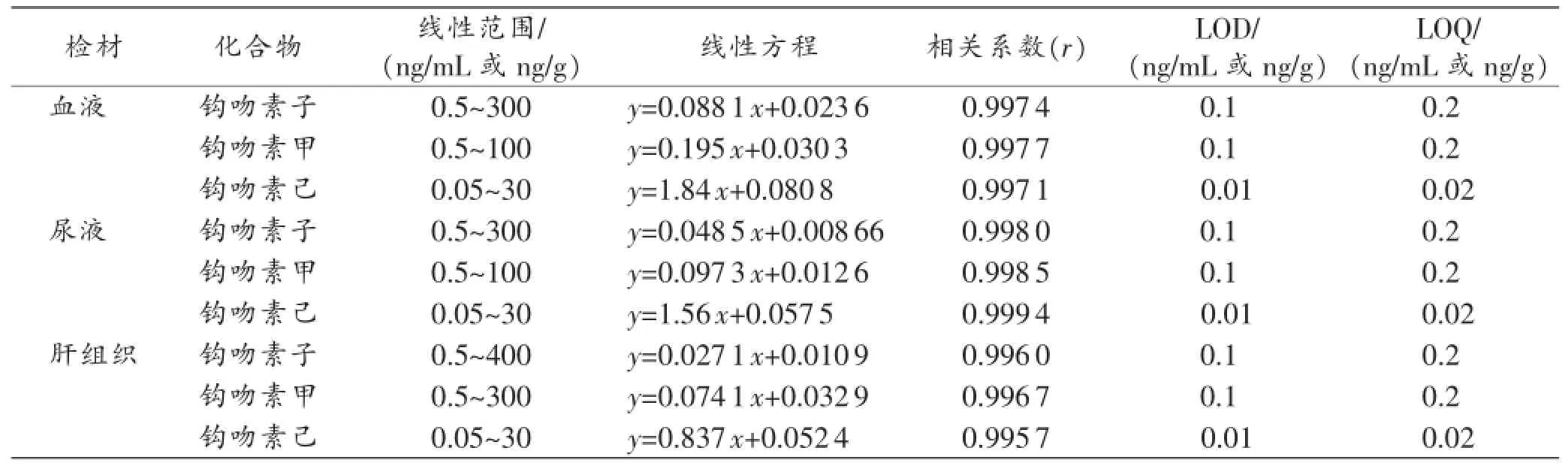
表2 生物检材中钩吻素子、钩吻素甲和钩吻素己的线性方程、相关系数(r)、LOD、LOQ
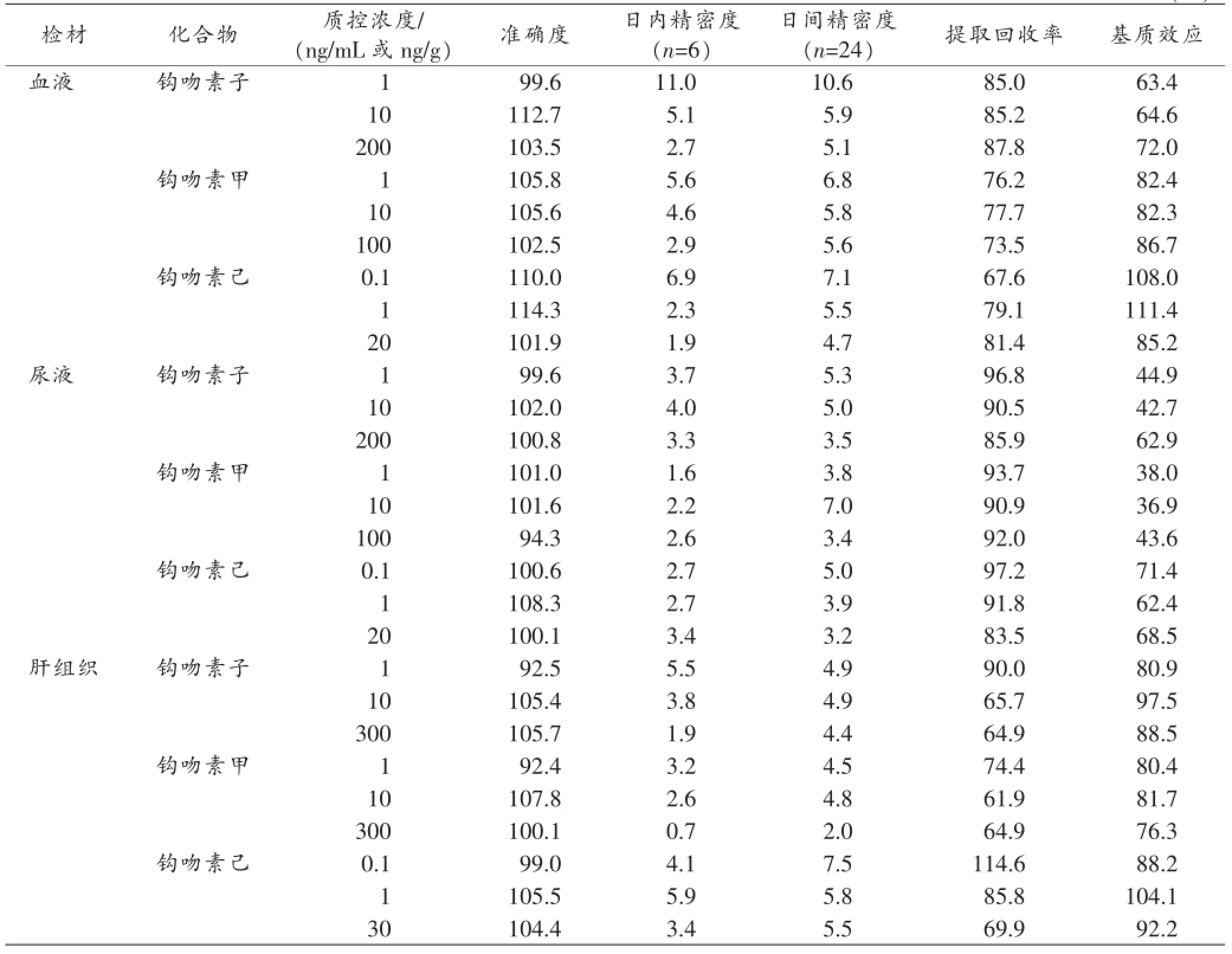
表3 各检材中钩吻素子、钩吻素甲和钩吻素己的准确度、精密度、提取回收率和基质效应(%)
2.2.5 稳定性
稳定性考察的结果显示,血液、尿液及肝组织样品在室温条件下放置24h、样品处理后于进样盘中放置24h、3次冻融循环(-20℃冰箱中放置21 h,转移到室温下放置3h,反复3次)、于-20℃冰箱中冷冻保存1个月这4种保存条件下,钩吻素子、钩吻素甲和钩吻素己相对偏差的绝对值均不超过19.6%,表明各待测物的稳定性良好。
3 结论
本研究建立了一种同时检测生物检材(血液、尿液和肝组织)中钩吻素子、钩吻素甲和钩吻素己的LCMS/MS法,该方法专属性强、灵敏度高,能为钩吻中毒的临床诊治和法医毒物分析提供有效的技术支持。
[1]黄静,苏燕评,俞昌喜,等.钩吻生物碱化合物体外抗消化系统肿瘤的活性[J].海峡药学,2010,22(3):197-200.
[2]周名璐,黄聪,杨小平.钩吻总碱的镇痛、镇静及安全性研究[J].中成药,1998,20(1):35-36.
[3]王友顺,高英立,刘上云,等.盐酸钩吻碱眼药水散瞳与调节麻痹作用的临床观察[J].中药药理与临床,1990,6(6):40-42.
[4]刘浩,俞昌喜.钩吻的研究进展[J].福建医科大学学报,2008,42(5):469-472.
[5]Jin GL,Su YP,Liu M,et al.Medicinal plants of the genus Gelsem ium(Gelsem iaceae,Gentianales)--a review of their phytochemistry,pharmacology,toxicology and traditional use[J].JEthnopharmacol,2014,152(1):33-52.
[6]孙莉莎,雷林生,方放治,等.钩吻素子对小鼠脾细胞增殖反应及体液免疫反应的抑制作用[J].中药药理与临床,1999,15(6):10-12.
[7]Liu M,Shen J,Liu H,et al.Gelsenicine from Gelsem ium elegans attenuates neuropathic and inflammatory pain in m ice[J].Biol Pharm Bull,2011,34(12):1877-1880.
[8]陈翼胜,郑硕.中国有毒植物[M].北京:科学出版社,1987:557-558.
[9]李磊,曾洋.利用钩吻自杀的法医学分析1例[J].广东公安科技,2015,23(2):74-75.
[10]许盈,郑宓,李苏平,等.钩吻素子在大鼠体内的药物代谢动力学及组织分布[J].福建医科大学学报,2013, 47(4):199-203.
[11]马克芹,王毅.GC/MS法测定人血中钩吻碱甲[J].微量元素与健康研究,2014,31(6):48-50.
[12]张春水,郑珲,何毅,等.钩吻素甲的气相色谱-质谱分析[J].质谱学报,2004,25(3):172-174.
[13]吴惠勤,张春华,黄晓兰,等.气相色谱-串联质谱法同时检测尿液中15种有毒生物碱[J].分析测试学报,2013,32(9):1031-1037.
[14]Zhang S,Hu S,Yang X,et al.Development of a liquid chromatography w ith mass spectrometry method for the determination of gelsemine in rat plasma and tissue:Application to a pharmacokinetic and tissue distribution study[J].J Sep Sci,2015,38(6):936-942.
[15]张春华,吴惠勤,黄晓兰,等.液相色谱-电喷雾串联质谱同时检测尿液和胃液中12种有毒生物碱[J].分析化学,2012,40(6):862-869.
[16]熊小婷,吴惠勤,黄晓兰.液相色谱-电喷雾串联质谱同时检测血液中8种有毒生物碱[J].分析化学,2009,37(10):1433-1438.
[17]Chen JZ,Li Y,Xiao JP,et al.Development of a sensitive and rapid UPLC-MS/MS method for the determ ination of koum ine in rat plasma:application to a pharmacokinetic study[J].Biomed Chromatogr,2013,27(6):736-740.
Simultaneous Quantitative Analysis of Koum ine,Gelsem ine and Gelsenicine in Biological Samp les by LC-MS/MS
JI Sheng-jie1,2,LIU Wei2
(1.Department of Forensic Medicine,School of Basic Medical Sciences,Fudan University,Shanghai 200032, China;2.Shanghai Key Laboratory of Forensic Medicine,Shanghai Forensic Service Platform,Institute of Forensic Science,Ministry of Justice,PRC,Shanghai 200063,China)
ObjectiveTo establish a LC-MS/MS method which is accurate and sensitive for determ ination of koum ine,gelsemine,and gelsenicine in biological samples and to verify the method.MethodsStrychnine was used as internal standard.Analytes in blood,urine and liver w ith 1%sodium hydroxide solution were extracted by ethyl acetate.Chromatographic separation was achieved on a ZORBAX SBC18column(150mm×2.1mm,5μm),and gradient elution was performed w ith the buffer solution of methanol-20mmol/L ammonium acetate(including 0.1%formic acid and 5%acetonitrile)as mobile phase. Qualitative and quantitative analysis was performed in the multiple reaction monitoring mode coupled w ith an electrospray ionization source under positive ion mode(ESI+).ResultsThe linearity of koumine, gelsemine and gelsenicine in blood,urine and liverwas good w ithin corresponding linear limitation and the correlation coefficients(r)>0.9950.The limits of detection were 0.1ng/m L(0.1ng/g),0.1 ng/m L(0.1ng/g)and 0.01ng/m L(0.01ng/g),respectively.The extraction recovery and accuracy of the alkaloids ranged from 61.9%to 114.6%and 92.4%to 114.3%,respectively.The relative standard deviations of the intra-day and inter-day precisions were not more than 11.0%.ConclusionThe method is selective,sensitive and suitable for simultaneous determination of koum ine,gelsemine and gelsenicine in body fluids and tissues, which offering technical support for clinical diagnosis and treatment and forensic toxicological analysis of Gelsemium elegans poisoning.
forensic toxicology;liquid chromatography-tandem mass spectrometry;koum ine;gelsem ine; gelsenicine;biological samples
DF795.1
:A
10.3969/j.issn.1004-5619.2017.02.007
1004-5619(2017)02-0141-07
2016-12-09)
(本文编辑:严慧)
十三五国家重点研发计划项目(2016YFC0800706);上海市法医学重点实验室资助项目(17DZ2273200);上海市司法鉴定专业技术服务平台资助项目(16DZ2290900)
姬圣洁(1991—),女,硕士研究生,主要从事法医毒物分析研究;E-mail:JSJ1990jsj@126.com
刘伟,女,主任法医师,硕士研究生导师,主要从事法医毒物分析研究;E-mail:liuw@ssfjd.cn
