气相色谱法测定经大孔树脂醇制备的二仙汤样品中残留有机溶剂
2017-04-18李燕思司梁宏
李燕思,鲁 毅,杨 波,司梁宏
药学
气相色谱法测定经大孔树脂醇制备的二仙汤样品中残留有机溶剂
李燕思,鲁 毅,杨 波,司梁宏*
气相色谱法;有机溶剂残留;二仙汤
二仙汤是张伯讷教授50年代创制的方剂,全方由温肾益精养血药仙茅、淫羊藿、八戟天、当归和滋阴泻火药知母、黄檗组成,主要用于更年期综合征的治疗和抗衰老,临床应用40余年,疗效确切,载入多部中医方剂学著作。近年来其药理活性不断有文献报道,而有关提取、洗脱工艺及质量标准的报道却较少。大孔树脂作为一种有效的分离纯化手段,运用于中药有效成分的提取分离中,并取得了良好的效果。但有不少人提出大孔树脂苯系列残留物安全性问题[1,2]。因此,为了考察经大孔树脂制备的中药二仙汤样品中残留物乙醇、正己烷、苯、甲苯、对二甲苯、苯乙烯和二乙烯苯等七种有机溶剂的安全性,采用气相色谱(GC)法进行测定。
1 材料与仪器
1.1 仪器岛津GC-14B气相色谱仪,FID检测器,N2000色谱工作站;Mettler AE 240型电子天平(瑞士梅特勒—托利多有限公司);KQ3200型超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药二仙汤样品(四五四医院自制);乙醇、正己烷、苯、甲苯、对二甲苯、苯乙烯、二乙烯苯、正庚烷、N,N-二甲基甲酰胺(DMF)均为分析纯。
2 方法与结果
2.1 色谱条件及系统适用性试验SupeLcoWAX10 30m×0.25mm×0.25μm毛细管柱,FID检测。载气为高纯氮(99.99%),分流比30∶1,进样量为0.2μl。毛细管柱初始柱温60℃,进样口温度180℃,检测器温度220℃。升温程序为60℃恒温1.1min,40℃/min升温至200℃,保持5min[3]。在上述色谱条件下,正己烷、乙醇、苯、甲苯、对二甲苯、苯乙烯和二乙烯苯色谱峰的理论塔板数不低于5000;且与内标物正庚烷的色谱峰分离度均>1.5,对照品及供试品的色谱图见图1。

图1 二仙汤GC色谱图
2.2 溶液配制
2.2.1 供试品溶液 精密称取醇提二仙汤原料药1.0 g,用DMF溶解并定容于10.0ml量瓶中,依法配制成待测供试品溶液。
2.2.2 对照品溶液 精密称取正己烷502.8mg、无水乙醇507.9mg、苯10.78mg、甲苯505.6mg、对二甲苯507.6mg、苯乙烯503.2mg和二乙烯苯502.6mg,以DMF溶解并定容至100ml量瓶中,作为标准对照品储备液;精密称取分析纯正庚烷509.1mg,以DMF溶解并定容于100ml量瓶中,作为内标物储备液。
2.3 线性关系考察精密吸取上述标准对照品储备液0.1、0.2、0.5、1.0、2.0和10.0ml,分别置于6个50ml量瓶,加入2.0ml内标物储备液,DMF定容至刻度,混匀,配制成标准溶液系列。依法进行测定,以待测组分与内标物的峰面积比(A)与对相应的待测组分浓度(C)进行线性回归,分别得标准曲线方程及线性范围:A正己烷=0.0083C-0.0456(r=0.9999),10.06~1006.0μg/ml;A乙醇=0.0051C+0.0574 (r= 0.9977),10.16~1016.0μg/ml;A苯=0.1183C+0.0342(r=0.9968),0.2156~21.56μg/ml;A甲苯=0.0115C+ 0.1705 (r=0.9975),10.11~1011.0μg/ml;A对二甲苯= 0.0116C+0.1556(r=0.9975),10.15~1015.0μg/ml;A苯乙烯=0.012C+0.1493(r=0.9975),10.05~1005.0μg/ ml;A二乙烯苯=0.0098C+0.1477 (r=0.9979),10.05~1005.0μg/ml。
2.4 检测限考察精密移取“2.3”项下的最低浓度标准溶液,通过不同比例稀释后进行测定,信噪比值约为3时,为最低检测限。正己烷检测限约为5.030μg/ml, 乙醇约为 5.080μg/ml, 苯约为0.1294μg/ml,甲苯约为5.055μg/ml,对二甲苯约为5.075μg/ml,苯乙烯约为5.03μg/ml,二乙烯苯约为5.540μg/m l。
2.5 精密度试验精密移取“2.3”项下的中间浓度标准溶液,按气相色谱测定条件连续进样5次,计算正己烷、乙醇、苯、甲苯、对二甲苯、苯乙烯、二乙烯苯和内标物正庚烷的峰面积比值,结果表明RSD分别为2.80%,3.48%,2.39%,3.97%,3.40%,4.83%,3.45%。
2.6 稳定性试验精密称取供试品原料药,按“2.3”项下方法制备供试品溶液,室温放置,分别于0、4、8、12、24 h按“2.1”项下色谱条件分析。计算正己烷、乙醇、苯、甲苯、对二甲苯、苯乙烯、二乙烯苯和内标物正庚烷的峰面积比值,结果表明RSD分别为 2.83%,2.52%,3.70%,3.16,%,0.97%,2.18%,3.24%。
2.7 加样回收率试验精密称取已知含量的供试品1.0 g,加入一定量的标准对照品储备液和内标储备液,按“2.2.1”项下方法配制成加样回收率溶液,进行测定,结果见表1。
2.8 含量测定按“2.2.1”项下方法制备供试品溶液,测定3个批次的有机溶剂残留量,见表2。

表1 加样回收率测定结果
3 讨论
3.1 溶剂的选择由于待测的有机溶剂种类多且含量少,导致峰面积较小,故先后摸索了几种溶剂,如水、DMF和DMSO。考虑二仙汤原料药难溶于水,大多数有机溶剂在水中溶解度小,在DMF、DMSO中溶解度较大,但DMSO对眼睛、皮肤等更具刺激性,且故采用DMF作为溶剂。
3.2 进样方式的选择顶空进样适用于容易挥发的样品[4],待定的有机溶剂种类较多,沸点差距较大;若采用顶空进样法,平衡时间较长[5,6],考虑检测效率,采用直接进样方式。
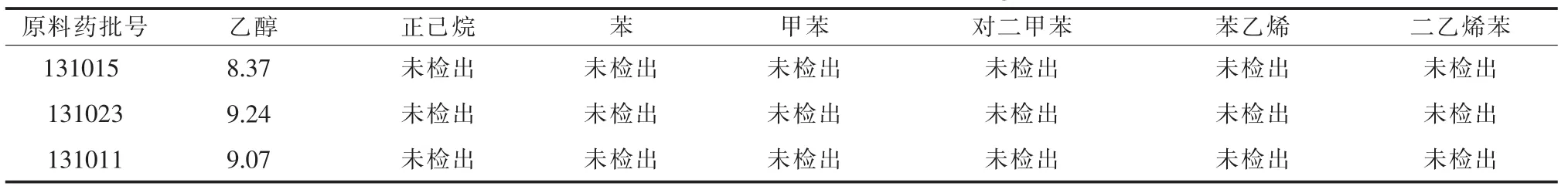
表2 醇提二仙汤原料药残留量测定结果(μg/m l)
3.3 程序升温速率的选择对不同程序升温速率(10、30、40、60、80℃/min)进行比较,发现升温速率越快,基线的漂移就越严重,乙醇峰和苯峰会出现重叠[7,8],为了缩短分析时间且各峰不相互干扰,最终选定40℃/min程序升温,使7种有机溶剂得到完全分离,60℃保持1min,让沸点低的溶剂正己烷和正庚烷(内标)析出,40℃/min升温至200℃,让乙醇、苯、甲苯、对二甲苯、苯乙烯、二乙烯苯依次分离,200℃保持5min,让沸点高杂质溶剂冲出柱子,以确保试验准确,选用正庚烷做内标不仅可以和待测组分有效分离,又能避免由于进样体积误差对测定结果的影响,从而获得比外标法更好的测定精密度和回收率[9]。
[1]高 青,张 喆,车宝泉.残留溶剂GC检查方法的设计与验证[J].中国药品标准,2007,8(3):10-13.
[2]武晓静,徐 慧,庄件兵,等.气相色谱法测定银杏内酯衍生物中有机溶剂残留量[J].中国野生植物资源,2008,27(6):67-69.
[3]李 薇,李小敏.头孢特仑新戊酯中有机溶剂残留量的毛细管GC测定[J].中国医药工业杂志,2007,38(10):729-730.
[4]蔡振华,金松子,蒋庆峰.顶空气相色谱法测定黄芪提取物中大孔树脂邮寄溶剂残留[J].现代仪器与医疗,2016,22(1):81-84.
[5]鲁晋南,王 丽,张国平,等.溶剂萃取-气相色谱法测定水果中二流代氨基甲酸盐类农药残留方法研究[J].中国卫生检验杂志,2016,26(19):2770-2772.
[6]孔祥虹,刘学芝,何 强,等.顶空气相色谱法测定植物提取物中 19中有机溶剂残留量[J].理化检测:化学分册,2013,49(8):924-930.
[7]张 超,马晓丽,谢登龙,等.气相色谱法测定乙二醛溶液中的乙二醇[J].山东化工,2016,17(45):72-74.
[8]曹忠波,赵婉婧,高 岩,等.固相萃取-气相色谱-质谱法检测水中16种邻苯二甲酸酯[J].中国卫生检验杂志,2016,26(18):2607-2610.
[9]霍建富,连建科,申云飞,等.气相色谱法测定人胎盘肝细胞生长素中有机溶剂残留量[J].山西医科大学学报,2016,47(4):348-351.
[2016-09-18收稿,2016-10-15修回]
[本文编辑:刘一洋]
R917
B
10.14172/j.issn1671-4008.2017.04.025
210002江苏南京,解放军454医院药学部(李燕思,鲁毅,杨波,司梁宏)
司梁宏,Email:1127153496@qq.com
