烃类热裂解动力学模型研究局限性及对策*
2017-03-15崔晓华张红梅任铭琪李金莲郝玉兰贺永殿
崔晓华,张红梅,任铭琪,林 枫,李金莲,郝玉兰,贺永殿
(1.东北石油大学 机械科学与工程学院,黑龙江 大庆 163318;2.东北石油大学化学化工学院 石油与天然气化工省重点实验室,黑龙江 大庆 163318;3.吐哈油田工程技术研究院,新疆 鄯善 838202)
目前,世界上98%的乙烯是由烃类蒸汽热裂解生产所得。由于不同的烃类结构所生成的乙烯、丙烯和丁二烯的数量变化很大[1],因此烃类结构成为影响乙烯工业生产成本的重要因素之一,特别是以石脑油等较重原料为主的企业。而建立可靠的烃类热裂解反应动力学模型对于优化裂解原料和相应裂解炉设计开发、裂解炉改扩建、延长在最佳工况条件下裂解炉的运转周期等都起着至关重要的作用,所以迫使人们不得不研究不同烃热裂解的动力学规律。动力学模型的建立,最早是用实验方法,根据实验结果进行分析、推导,得到其自由基机理;由于工业生产的需求,也建立了如集总动力学、分子反应动力学等模型;一些用计算机模拟研究动力学的新方法也在不断出现[2-6],但研究精度仍是一个难以解决的关键问题。也就是说,到目前为止,真正精度高、模拟计算结果准确、能适应我国裂解的原料种类增多、不同原料切换时预测评价精度的动力学模型并不多。作者对我国热裂解动力学模型的研究现状进行了分析,并根据研究结果提出了相应对策。
1 裂解反应动力学模型研究的误区及出路
热裂解反应动力学模型研究的主要困难如下。
(1) 单一烃裂解自由基反应机理的复杂性。虽然Rice[7]在1931年就用实验方法确定了热裂解反应是按照自由基机理进行的,但迄今为止实验方法仍不能确定较大分子烃类热裂解的反应机理,对单一烃的研究文献主要集中在C10以内的烷烃,而对于其自由基机理研究较清晰的烷烃主要集中在C4以下,说明了实验研究的困难;
(2) 原料组成的复杂性。工业裂解原料一般是由不同烃类组成的混合物,实验结果表明,不同烃所生成的自由基不同,且这些自由基会产生一定的相互作用,从而使裂解产物分布与单独裂解时最终产物不同;
(3) 裂解反应过程的复杂性。裂解反应是强吸热反应,反应温度高,停留时间短,且在小口径管式反应器中进行着包括流体流动、传热、裂解反应等复杂过程,由于这些过程之间具有高度的耦合性,管内的流动状态及反应规律不易测得,要直接了解管内裂解反应的详细规律以及测定反应器内的一些工艺参数是极其困难的。
综上所述,正是由于烃类自由基机理实验研究时多重复杂性的叠加,才导致了许多研究者避开了对反应机理过程的研究,而把精力放在了从原料组成和产物分析数据来回归动力学规律这一方法上,因此出现了相对比较简单的分子模型、集总模型等动力学研究方法,也恰恰是由于重点集中在了对实验原料、反应结果的研究及人为推测上,忽略了对自由基反应过程规律和特点的重视,才是这些模型只能适应特殊的原料组成,且无法进一步改进方法的最根本原因。因为动力学规律恰恰应该由原料进行反应的过程中去寻找,也就是说,反应的本质规律和特点的掌握才是动力学模型准确性提高的关键。而根据Rice提出的自由基机理来建立的各个自由基基元反应由于独立于原料、裂解装置以及裂解工艺条件之外,因此决定了该模型具有良好的适应性与外延性,是用于描述和总结烃类蒸汽热裂解反应规律的理想模型。因此要想提高动力学模型的准确性和外延性,必须从机理模型入手。
2 自由基机理模型的研究现状及困难
对单一烃类分子进行自由基机理研究一般采用如下的方法:首先对该烃类分子进行实验,通过对实验结果的分析及总结,得到该分子进行热裂解可能发生的自由基反应,并通过实验测定或计算这些自由基反应的活化能和指前因子;然后以上述实验数据为动力学模型,假设在反应达到稳态时各自由基产生和消失的速度相等,得到不同产物及自由基的非线性微分方程组;最后用微分方程的数值解法对非线性方程组进行求解,得到各产物及自由基沿管长的变化规律。张红梅等[8]对乙烷热裂解反应机理进行了数值模拟理论研究,采用Materials Studio(MS)分子模拟软件对乙烷热裂解可能发生的主要反应进行模拟计算,计算结果见表1。

表1 乙烷热裂解自由基反应及动力学参数
将上述自由基反应进行任意组合,可设计得到不同的4个反应路径。按文献[9]中 “假设在反应达到稳态时各自由基产生和消失的速度相等”的方法进行推导,各路径的活化能(Ea)见表2。

表2 乙烷热裂解不同反应路径的活化能推导值
根据文献[9]中的实验数据可知,实际测得乙烷热裂解的反应活化能为264~294 kJ/mol,上述结果都与实验结果不符。为了解释上述方法计算结果为何与实验数据不符的原因,作者采用Aspen软件建立了管式炉反应器进行了模拟计算,结果发现,上述动力学模型在Aspen管式炉反应器进行反应的过程,与该推导方法有如下明显不同[8]。(1)在实际的反应过程中,只有在反应开始时有少量的链引发反应,产生少量自由基后大量进行的是链传递和链循环反应,由于自由基的摩尔分数一般在约10-6,因此链引发和链终止反应对反应路径活化能的影响很小,而按文献[9]的方法上述4个反应路径1~3只是链终止反应不同,但得到的反应路径的活化能却相差很大,且都与实验结果不符;(2)每个路径中都会出现不同的小路径,只有当某一路径产生的自由基能形成循环反应时,才可能影响到产物分布,而上述推导只是根据活化能进行组合;(3)在原料消耗的不同阶段,反应会发生一定变化的,而不是机会均等。由此可见,按文献[9]的方法进行推导存在着配平的任意性,且不同的配平方法将得到截然不同的结果,这是经典数值方法的局限性。“假设在反应达到稳态时各自由基产生和消失的速度相等”,是由于链循环产生了同样的自由基,导致了自由基数量的相对不改变,这与链终止反应并无关系。正是这一方法存在的任意性和人为因素使得计算结果与实验结果不符,影响了对烃类热裂解机理的研究。
3 自由基机理模型研究方法的改进
随着计算机技术的迅速发展,分子模拟等数值模拟技术的功能越来越强大,被广泛应用到多个研究领域。张红梅等[8,10-14]首先将分子模拟技术与已有的烃类裂解规律相结合研究烃类热裂解机理,运用该技术初步研究了C5以下单一烃的热裂解自由基反应动力学数据,进行了模拟计算,得到了与实验结果接近的主要产物分布,验证了分子模拟的可行性。在对现有自由基机理研究方法局限性进行分析的基础上,提出了将分子模拟和管式反应器工艺过程模拟相结合模拟计算烃类热裂解自由基反应机理的理论方法,并将文献中有关烃类热裂解的实验研究结果与分子模拟和工艺模型研究相结合,改进了现有实验和理论研究方法的不足。
3.1 对实验数据的校正及对实验方法的改进作用
裂解实验数据一般是由单一或多个原料烃组成的原料所引发的复杂反应网络进行自由基反应后得到的结果,有些起决定性作用的反应可以通过实验数据的分析得到准确的结果,这些裂解规律目前已经熟知。但由于裂解反应的特殊性,研究者在对实验结果进行推导时对一些细节性的问题往往会有个人臆想的因素存在,使得不同研究者研究结果各不相同。这也是目前动力学模型准确性低的一个原因。而采用分子模拟计算可以对实验数据进行校正或通过对实验方法的改进,可得到更准确的动力学数据。丙烷热裂解自由基反应的活化能数据、实验数据、分子模拟计算结果及两者的相对误差见表3。
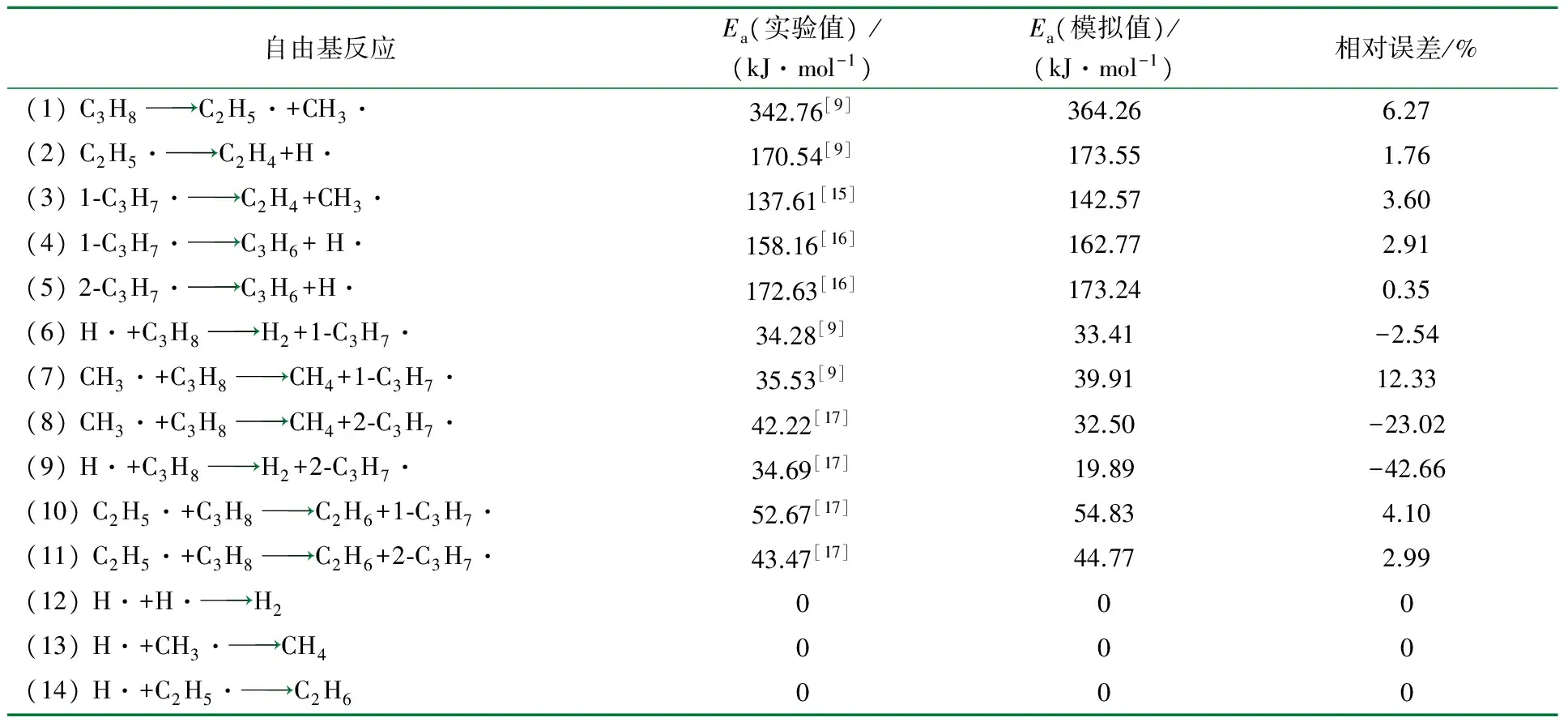
表3 丙烷热裂解自由基反应及动力学参数
由模拟数据可以看出,丙烷的链引发反应的活化能最大,其次是较大自由基的分解反应,活化能次之,活泼自由基链传递反应的活化能最小,模拟计算结果符合已有的裂解规律。由相对误差可以看出,大多数模拟计算值与文献值是符合的,只有活化能较小、反应较快的自由基链传递反应(表3中的反应7、8和9)有较大的相对误差。通过对比分析表3中活化能相差较大的原因如下。
(1) 模拟计算得到的氢和甲基自由基与乙烷、丙烷和正丁烷进行链传递反应时的活化能数据与文献值的对比见表4。从表4可以看出,对于同一反应,不同文献所提供的实验或理论计算值本身就存在着较大的差别,充分说明了自由基链传递反应活化能测定的困难及实验过程控制的难度。根据已有的烃类热裂解规律,氢或甲基自由基分别夺取乙烷、丙烷与正丁烷伯碳上的氢时,所需活化能应该是依次增大,分子模拟结果符合这一规律,而文献数据中的甲基自由基不符合这一规律。说明自由基变大时,实验难度增大,实验数据误差也随之增大,而模拟计算值能更好地与这一规律吻合;

表4 氢自由基、甲基自由基与小分子烷烃链传递反应活化能的实验数据
(2) 根据Aribike[17]、杨毓溥等[18]的研究成果可知,同一自由基与烃类分子进行链传递时,被传递的烃类分子越大,反应所需的活化能越大;被传递的烃类分子不变时,随着自由基碳原子数的增加,传递反应所需的活化能也逐渐增大。由表4可知,模拟计算结果中氢自由基夺取乙烷、丙烷和正丁烷伯碳上的氢时的活化能模拟计算值依次增大;甲基自由基攻击乙烷、丙烷和正丁烷的伯碳时的活化能模拟计算值也依次增大,且氢自由基夺取同一烷烃相同位置时的活化能均小于甲基自由基,与上述文献研究结果完全吻合,而实验数据的吻合程度要明显低于模拟数据;
(3) 从表4的数据还可以看出,氢自由基和甲基自由基分别夺取伯碳上的氢时活化能大于仲碳上的氢,与烃类分子键能中间小、两边大的结论相吻合;而实验对于这些细节问题的研究就比较困难。
综上所述可以看出,分子模拟不仅能得到符合裂解规律的计算结果,还可以对实验测定比较困难的一些自由基反应给出更精确、合理的解释,可以作为研究热裂解反应的一个手段。
3.2 对实验解决不了的细节问题的处理
由于自由基具有反应速率快、存在时间短、数量巨大的特点,因此利用实验很难观察到一些细节问题,而理论研究却可以弥补这一不足。例如,由分子模拟可知正丁烷、正戊烷和正己烷的C—H键键长的数据见表5(表中①②③表示碳的位置)。

表5 三种正构烷烃的C—H键的键长 键长/pm
由表5数据可知,伯碳的C—H键长最短,最不易断裂;仲碳上的C—H键长都大于伯碳的C—H键长,更容易断裂,且越靠近分子中央的仲碳越易断裂,这是符合一般裂解规律的。由分子模拟得到的这3个正构烷烃夺氢反应的活化能数据见表6。
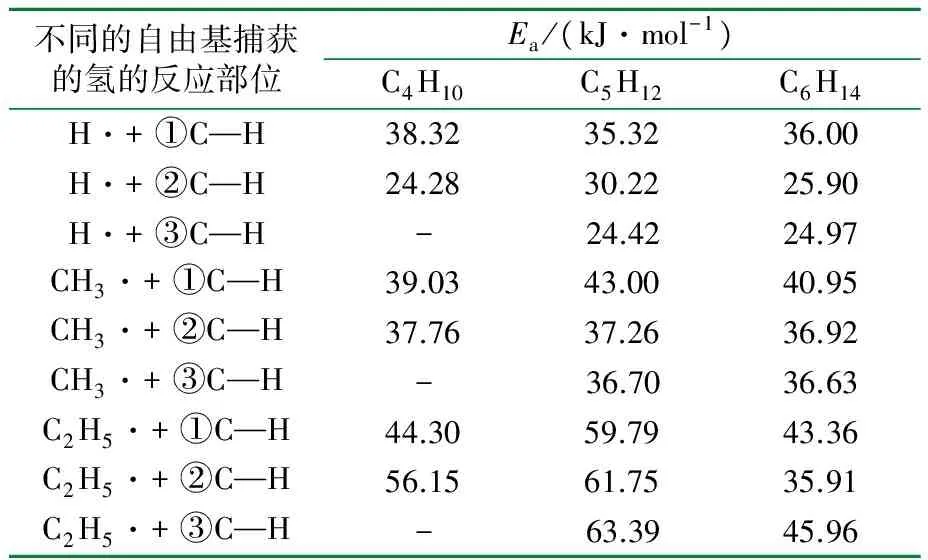
表6 三种正构烷烃C—H键断裂的活化能
由表6可知,H·和CH3·与正戊烷进行夺氢链传递反应的活化能数据与键长数据吻合,但C2H5·夺取仲氢时的活化能却是仲碳大于伯碳。如果仅从键长的角度看,各个自由基在夺取仲氢时所需要的活化能应该比较小,但是,在实际发生反应的过程中,还应考虑自由基大小所引起的空间位阻影响。H·和CH3·本身基团比较小,因此空间位阻的影响较小,因此其活化能的数据符合已有的裂解规律;而C2H5·基团较大,与仲氢原子发生反应时,会因与周围的氢距离太近,进入排斥区域,从而使得C2H5·夺氢的空间位阻增大,使得活化能变大;另外还要注意靠近正构烷烃两端倒数第二个碳的特殊位置。由正己烷的结构图(见图1)可知,正己烷的②号碳和⑤号碳虽然也是仲碳,但由于紧靠两端的①号和⑥号碳上氢连接方式的改变,可使其空间位阻减少,因此其活化能较小,而靠近中间的③、④号碳和正戊烷的③号碳上的氢具有更紧密的排列结构,使得空间位阻增大,从而导致了活化能的增加。正丁烷因不存在这样的结构,故所有计算结果都符合裂解规律。
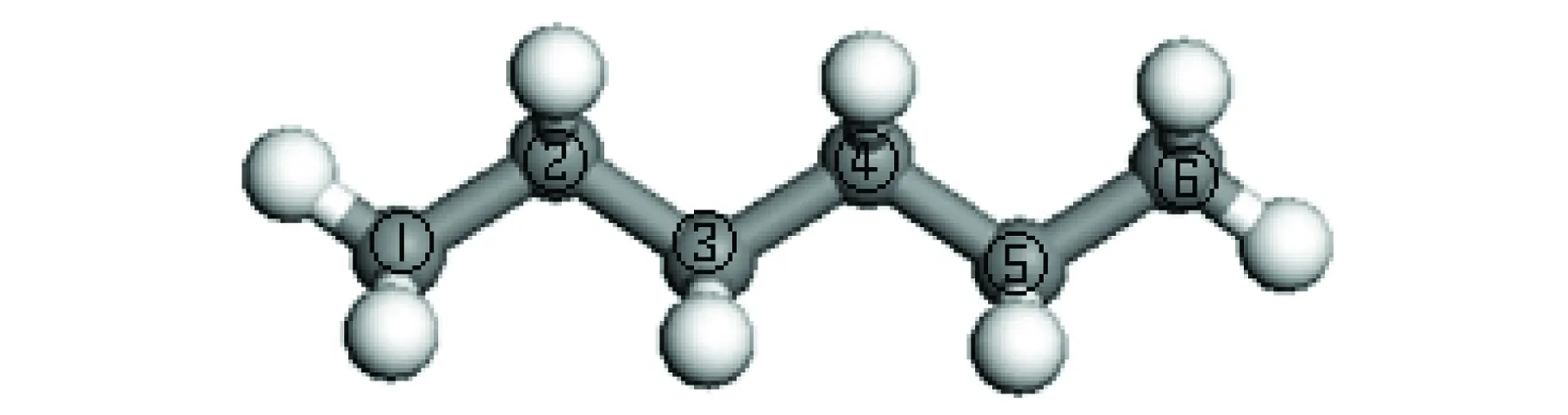
图1 正己烷结构图
4 结束语
(1) 通过对目前已有的蒸汽热裂解动力学模型的分析,认为经验模型、集总模型和分子模型只是通过原料组成和最后产物分布数据建立模型,而忽略了对反应过程的详细研究,而反应过程才是影响模型准确性最关键的因素,因此注定了该类模型具有外延性差的缺点,要想得到外延性好的模型必须从机理模型入手;
(2) 目前已有的机理模型的实验研究方法存在的困难使得对热裂解机理模型的研究进展缓慢,不适应目前我国乙烯工业的发展,且存在细节性实验数据不准、整体数据规律性差的缺点,而计算机模拟这一新兴理论研究方法虽然研究速度快、省时省力,但目前还未得到人们的广泛认可;
(3) 在对现有自由基机理实验方法的局限性进行分析的基础上,提出了将分子模拟、管式反应器工艺模拟的理论方法与实验研究相结合的研究烃类热裂解自由基反应机理的新思路,该方法可以弥补实验和理论方法各自的不足,从而较准确、快速地研究烃类热裂解的自由基反应机理,为高精度的通用模型的建立提供大量有用的信息。
[1] 万书宝, 贺德福.蒸汽裂解制乙烯的发展趋势[J].现代化工,2009,29(6):6-10.
[2] ALEJANDROO DOMANCICH,VIRGINIA PEREZA,PATRICIA M HOCH,et al.Systematic generation of a CAPE-OPEN compliant simulation module from GAMS and FORTRAN models[J].Chemical Engineering Research and Design,2010,88(4):421-429.
[3] ZHAO Y X,ZHANG S J,LI D.Understanding the mechanism of radical reactions in 1-hexene pyrolysis[J].Chemical Engineering Research and Design,2014,92(3):453-460.
[4] ZHANG Y J,CAI J H,ZHAO L,et al.An experimental and kinetic modeling study of three butene isomers pyrolysis at low pressure[J].Combustion and Flame,2012,159(3):905-917.
[5] 沈本贤,田立达,刘纪昌.基于结构导向集总的石脑油蒸汽裂解过程分子尺度动力学模型[J].石油学报(石油加工),2010(增刊1):218-225.
[7] RICE F O.The thermal decomposition of organic compounds from the standpoint of free radicals.Ⅰ.Saturated hydrocarbons[J].Journal of the Americian Chemical Society,1931,53(5):1959-1972.
[8] 张红梅,李青月,李金莲,等.乙烷丙烷单独及混合裂解相互作用机理的模拟研究[J].化工科技,2015,3(1):9-13.
[9] 吴指南.基本有机化工工艺学[M].北京:化学工业出版社,1990:23-30.
[10] 张红梅,张晗伟,顾萍萍,等.异丁烷热裂解反应机理的分子模拟[J].化工学报,2012,63(10):3138-3142.
[11] 张红梅,顾萍萍,张晗伟,等.丙烷热裂解反应机理的分子模拟[J].石油学报(石油加工),2012,28(6):986-990.
[12] 郝玉兰,张红梅,张晗伟,等.丁烷热裂解反应机理的分子模拟[J].石油学报(石油加工),2013,29(5):68-73.
[13] 张红梅,温静,张晗伟,等.丙烷热裂解反应机理及路径的研究[J].青岛科技大学学报(自然科学版),2014,35(2):129-132.
[14] 张红梅,孙维,李金莲,等.正戊烷热裂解一次反应机理的数值模拟[J].石油学报(石油加工),2016,32(2):394-400.
[15] 倪力军,张立国,倪进方,等.链烷烃热裂解过程结构动力学模型与模拟[J].化工学报,1995,46(5):562-570.
[16] 奥尔布赖特.裂解理论和工业实践[M].北京:烃加工出版社,1990:53-65.
[17] ARBIKE D S,SUSU A A.Mechanistic modeling of the pyrolysis ofn-heptane [J].Thermochimica Acta,1988,127(16):259-273.
[18] 杨毓溥,刘德泉.对自由基交换反应中某些关系式的探讨[J].河北大学学报,1964(3):137-152.
