氨基酸型两性膦配体-铑催化剂催化高碳烯烃均相氢甲酰化反应规律的研究*
2017-03-15马青青徐冰莹曲俊蓉于世涛
金 欣,马青青,徐冰莹,曲俊蓉,于世涛
(青岛科技大学 化工学院,山东 青岛 266042)
铑催化的碳碳双键的氢甲酰化反应是一类典型的原子经济反应,根据底物的不同可以制备各种类型的醛和醇,在制药和精细化工等许多领域有着广泛的应用[1-2]。与所有的均相催化反应一样,氢甲酰化反应工业化过程中遇到的主要困难是贵金属催化剂与产物的分离问题[3-4]。
为了解决贵金属催化剂的回收与循环使用问题,水/有机两相催化的概念被提出。1984年,Ruhrchemie公司和Rhone-Peoulenc公司率先将水溶性膦配体TPPTS(三苯基膦三间磺酸钠盐)应用于铑催化的丙烯的水/有机两相氢甲化制备丁醛的生产中[5],随后大量的水溶性膦配体被相继开发出来。目前,常用的水溶性膦配体依据亲水基团的不同可分为阴离子型[6]、阳离子型[7]、非离子型[8]和两性离子型[9-13]四类,相比前三类水溶性膦配体,两性离子型水溶性膦配体的研究相对较少。
Fell[10]首次合成了一类结构中含有季铵基和磺酸基的两性膦配体,并应用于烯烃的水/有机两相氢甲酰化反应,具有较高的催化活性。Brauer等[11]设计合成出苯环上键连甘氨酸侧链的三苯基膦型配体。Heinicke等[12]合成出一类磷原子直接链接到氨基酸上的单膦配体,该配体被应用于乙烯齐聚反应中,α-烯烃选择性达到90%。Herrmann等[13]用三羟甲基膦和各种α-氨基酸合成了一系列两性水溶性单膦配体,并用于丙烯的水/有机两相氢甲酰化反应中,丙烯转化率达到93%,但催化剂稳定性较差,只循环使用了3次。
作者以Stelzer等设计的三苯基膦型氨基酸两性膦配体为模型(见图1),详细研究了该类配体的铑催化剂在催化高碳烯烃氢甲酰化反应中的规律,为两性膦配体在催化反应中的应用提供了一定的理论支持。
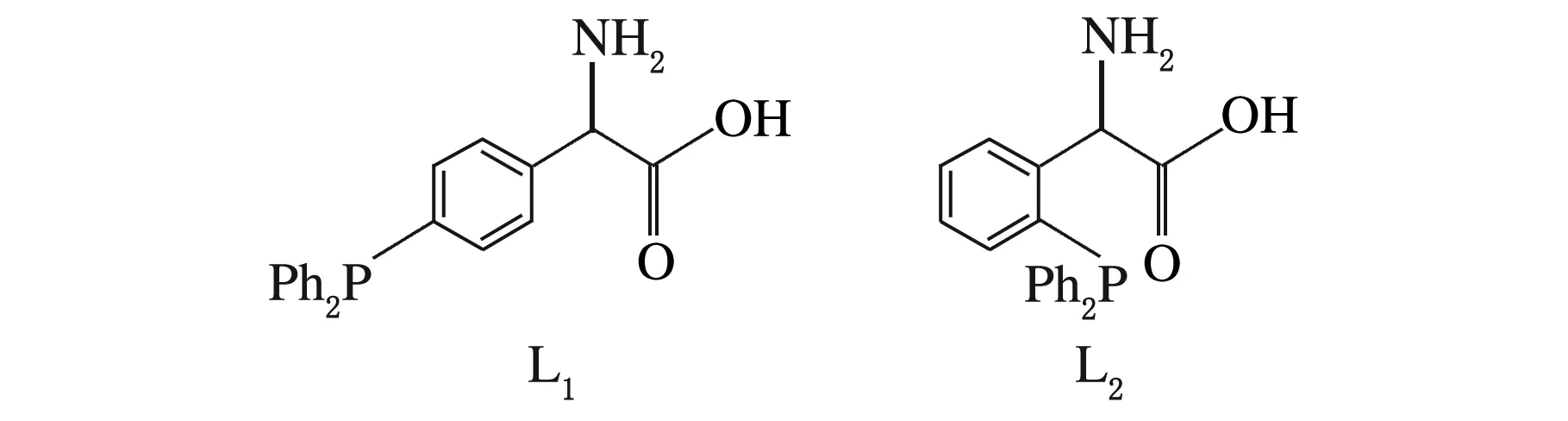
图1 基于三苯基膦的氨基酸型两性膦配体
1 实验部分
1.1 试剂与仪器
三水合三氯化铑:陕西开达化工有限责任公司;1-辛烯:德国ABCR公司;1-癸烯、1-十二烯、1-十四烯、四丁基氢氧化铵(质量分数25%的甲醇溶液):上海晶纯试剂有限公司;十二烷基苯磺酸:日本梯希爱化成工业发展有限公司;三乙胺:天津市博迪化工有限公司;膦配体L1和L2按照文献[11]合成;合成气[V(H2)∶V(CO)=1∶1]:青岛合利气体有限公司;以上所用试剂均为分析纯,溶剂经无氧无水处理后使用,实验操作均在Schlink真空线上进行。
不锈钢高压反应釜:50 mL,大连理工大学;气相色谱仪:SP2100A,OV101毛细管柱(50 m×0.25 mm I.D),北京北分瑞利分析仪器(集团)有限责任公司;气质联用仪:HP5890-HP5989A,DB-5毛细管柱,日本惠普公司。
1.2 烯烃的氢甲酰化反应过程
在N2的保护下,在高压反应釜内加入RhCl3·3H2O、膦配体(L1或L2)、添加剂、烯烃、内标(环己烷)和溶剂,用合成气置换体系6~7次,然后充至反应所需压力,在设定的温度反应一定的时间。取出反应釜在冰浴中快速降温至室温,将高压釜中的气体放空,取反应液进气相色谱仪分析,内标法定量。
2 结果与讨论
2.1 配体结构、溶剂和相转移催化剂对1-辛烯氢甲酰化反应的影响
氨基酸型两性膦配体的结构、溶剂和相转移催化剂对铑催化的1-辛烯氢甲酰化反应的影响见表1。从表1可以看出,以甲苯作为溶剂的情况下,当应用配体L1时,1-辛烯的转化率并不高,只有30%(序号1)。通过仔细观察发现,极性较强的氨基酸型两性膦配体L1在极性较弱的甲苯中的溶解性不佳,导致烯烃的转化率下降。当加入相转移催化剂十二烷基苯磺酸(C12PhSO3H)后,由于C12PhSO3H与两性膦配体上的氨基成盐,配体L1的溶解性变好,但1-辛烯的转化率反而进一步降至8.4%(序号2),产物醛的选择性也大幅下降,生成大量异构化烯烃。这一现象主要与三氯化铑前体和配体在合成气作用下形成铑催化活性物种的机理有关。按照Wilkinson催化剂的形成机理[14],以三氯化铑作为前体形成Wilkinson催化剂的同时有盐酸生成,对氢甲酰化反应会产生抑制作用。由于加入的相转移催化剂C12PhSO3H显强酸性,强烈抑制了铑催化活性物种的形成,使铑催化剂的活性大幅下降。为了验证这一判断,在加入C12PhSO3H的同时加入一定量的三乙胺使催化体系呈弱碱性[15],1-辛烯的转化率和醛的选择性均大幅提高,1-辛烯近于完全转化(序号3)。鉴于这一结果,在催化体系中直接引入碱性相转移催化剂四丁基氢氧化铵(NBu4OH),由于NBu4OH与两性配体的羧基成盐,配体的溶解性增强,催化活性和选择性均能保持在较高的水平(序号4)。

表1 配体结构、溶剂和酸碱添加剂对1-辛烯氢甲酰化反应的影响1)
1)t=80 ℃,p=2.0 MPa,t=2 h,n(配体)∶n(Rh)=5,n(1-辛烯)∶n(Rh)=1 000,m(RhCl3·3H2O)=1.0 mg,V(溶剂)=1.5 mL;2) 正构醛与异构醛的物质的量比。
研究发现,氨基酸型两性配体的结构也对氢甲酰化反应的活性产生较大影响。甘氨酸处于邻位的配体L2基本没有催化活性,其主要原因可能是配体L2的空间位阻较大,影响了其与铑形成有效的配位,导致催化剂失活。随后,采用强极性的甲醇替代弱极性的甲苯作为溶剂,由于配体L1在甲醇中的溶解性较好,即使不加入相转移催化剂,仅加入三乙胺,烯烃也接近完全转化(序号6)。在给出的所有条件下,正异比均在2.5~3.0。由于氨基酸型两性膦配体L1的母体结构是三苯基膦(TPP),在表1中TPP被作为参比配体进行了考察。在无任何添加剂的情况下,采用TPP时,烯烃转化率不到1%(序号7),远低于使用L1配体时的30.3%,即使加入三乙胺,烯烃转化率只提高至7.9%(序号8)。这一对比结果说明,氨基酸配体上的氨基能够中和生成铑活性物种时产生的盐酸,从而提高铑催化剂的催化活性。
2.2 反应温度对1-辛烯氢甲酰化反应的影响
反应温度对L1-Rh催化的1-辛烯氢甲酰化反应的影响见表2。

表2 反应温度对1-辛烯氢甲酰化反应的影响1)
1)p=5.0 MPa,t=4 h,n(L1)∶n(Rh)=5,n(1-辛烯)∶n(Rh)=1 000,m(RhCl3·3H2O)=1.0 mg,V(甲苯)=1.5 mL,n(L1) ∶n(C12PhSO3H) ∶n(NEt3)=1∶1∶10。
从表2可以看出,在其它反应条件相同的情况下,温度为60 ℃时,烯烃的转化率较低,仅为68.6%;当温度升高至80 ℃后,1-辛烯的转化率和醛的选择性均达到98%以上,继续提高反应温度,烯烃转化率几乎无明显的增加,而醛的选择性反而略有下降,这与反应温度升高后,烯烃的异构化副产物增加有关。此外,随着反应温度的升高,产物醛的正异比略有下降,说明高温有利于异构醛的生成,主要原因可能是异构醛的空间位阻大于正构醛,所以生成异构醛的活化能较高,因此较高的反应温度有利于异构醛的生成,导致正异比下降。
2.3 反应压力对1-辛烯氢甲酰化反应的影响
合成气压力对L1-Rh催化的1-辛烯氢甲酰化反应的影响见表3。
由表3可以看出,反应压力从1.0 MPa升至2.0 MPa后,1-辛烯转化率有较大程度的提高,从22.2%提高至98.1%,继续增加压力,烯烃转化率和醛的选择性均无明显增加。但是,随着压力的增加,CO的分压也增加,更有利于CO与铑配位形成空间位阻较小的HRh(CO)2(L1)2(见图2),从而更有利于异构醛的生成,导致正异比略有降低。

表3 反应压力对1-辛烯氢甲酰化反应的影响1)
1)t=80 ℃,t=2 h,n(L1)∶n(Rh)=5,n(1-辛烯)∶n(Rh)=1 000,m(RhCl3·3H2O)=1.0 mg,V(甲苯)=1.5 mL,n(L1) ∶n(C12PhSO3H) ∶n(NEt3)=1∶1∶10。
图2铑催化活性物种的转化平衡
2.4 反应时间对1-辛烯氢甲酰化反应的影响
反应时间对L1-Rh催化的1-辛烯氢甲酰化反应的影响见表4。
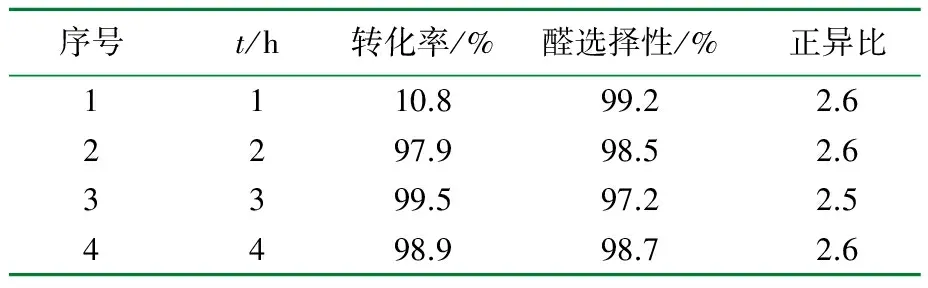
表4 反应时间对1-辛稀氢甲酰化反应的影响1)
1)t=80 ℃,p=5.0 MPa,n(L1)∶n(Rh)=5,n(1-辛烯)∶n(Rh)=1 000,m(RhCl3·3H2O)=1.0 mg,V(甲苯)=1.5 mL,n(L1) ∶n(C12PhSO3H) ∶n(NEt3)=1∶1∶10。
由表4可以看出,反应时间从1 h延长至2 h,烯烃转化率从10.8%提高至97.9%,继续延长反应时间,烯烃转化率变化不大,而正异比始终保持相对稳定。
2.5 不同烯烃的氢甲酰化反应
不同的烯烃底物的氢甲酰化反应见表5。
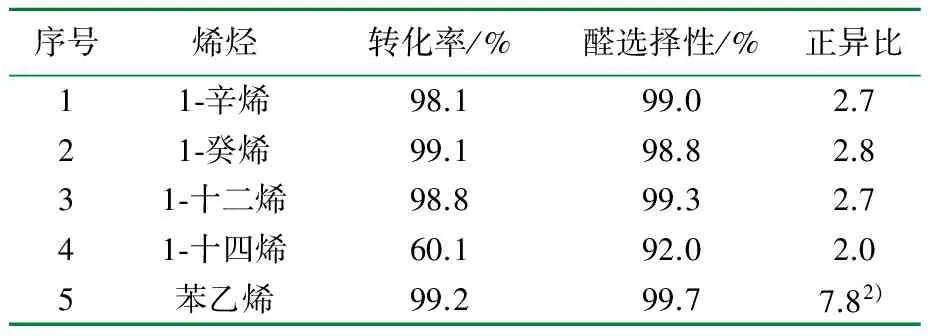
表5 不同烯烃的氢甲酰化反应1)
1)t=80 ℃,p=2.0 MPa,t=2 h,n(L1)∶n(Rh)=5,n(1-辛烯)∶n(Rh)=1 000,m(RhCl3·3H2O)=1.0 mg,V(甲苯)=1.5 mL,n(L1) ∶n(C12PhSO3H) ∶n(NEt3)=1∶1∶10;2) 此处为异正比值。
从表5中可以看出,从1-辛烯到1-十二烯,烯烃的转化率、醛的选择性和正异比基本变化不大。而对于1-十四烯,转化率下降幅度较大,只有约60%,正异比也有明显降低。这是由于随着烯烃的碳链增长,反应活性下降所致。而对于苯乙烯,除保持高的转化率和醛的选择性外,其主产物是异构醛,而非正构醛,异正比达到7.8。
3 结 论
系统研究了氨基酸型两性膦配体的结构、溶剂、相转移催化剂、反应温度、反应压力和反应时间对烯烃氢甲酰化反应规律性的影响,并考察了催化反应体系对不同底物烯烃的适用性。结果表明,膦配体的结构对催化活性具有较大影响,邻位带有氨基酸侧链的配体L2受到空间位阻的影响,活性较低;而对位有氨基酸取代的膦配体L1催化活性不受影响。以弱极性的甲苯作为溶剂时,添加碱性相转移催化剂四丁基氢氧化铵能够有效提高配体L2的溶解性和催化活性。反应温度,压力和反应时间对烯烃转化率和醛选择性的影响都存在一个突变点,超过突变点后,在较低的温度和压力下烯烃的转化率和产率都会有大幅度的提高,而且继续升高温度、压力和延长时间,转化率和选择性不会有明显的变化。此外,氨基酸两性配体对不同的烯烃底物也表现出较好的适用性。与三苯基膦配体相比,氨基酸型两性膦配体的耐酸碱性较强,在较宽的pH值范围内均表现出较高的催化活性,表明氨基酸型两性膦配体是一类性能优异的新型配体。
[1] BAHRMANN H,BOGDAMOVIC S.Aqueous-phase organometallic catalysis[M].Weinheim:Wiley-VCH,1998:306-321.
[2] THOMAS S,VORHOLT A J,ARNO B.The mission of addition and fission-catalytic functionalization of oleochemicals[J].European Journal of Lipid Science & Technology,2015,118(1):3-25.
[3] CORNILS B,HERRMANN W A.Applied homogeneous catalysis with organometallic compounds[M].Weinheim:Wiley-VCH,1996:29.
[4] POGRZEBA T,MÜLLER D,HAMERLA T,et al.Rhodium catalysed hydroformylation of long-chain olefins in aqueous multiphase systems in a continuously operated mini-plant[J].Industrial & Engineering Chemistry Research,2015,48:54-59.
[5] KUNTZ E G.Homogeneous catalysis in water[J].Chemtech,1987,17:570-575.
[6] SIEFFERT N,WIPFF G.On the importance of the aqueous interface in the multiphasic rhodium catalyzed hydroformylationof propene:a molecular dynamics study[J].Journal of Physical Chemistry C,2008,112(38):14891-14901.
[7] BRASSE C C,ULLIENGLERT A,SALZER A,et al.Ionic phosphine ligands with cobaltocenium backbone:novel ligands for the highly selective,biphasic,rhodium-catalyzed hydroformylation of 1-octene in ionic liquids[J].Organometallics,2000,19(19):3818-3823.
[8] JIN Z,ZHENG X,FELL B.Thermoregulated phase transfer ligands and catalysis.I.Synthesis of novel polyether-substituted triphyenylphosphinesand application of their rhodium complexes in two-phase hydroformylation[J].Journal of Molecular Catalysis A Chemical,1997,116:55-58.
[9] 金欣,刘国兵.基于PPM的氨基酸型两性水溶性手性膦配体前体的合成[J].现代化工,2010,30(1):68-70.
[10] FELL B,PAPADOGIANAKIS G.Rhodiumkatalysierte mizellare zweiphasenhydroformyIierung vonn-tetradecen-1 mit grenzfliihenaktiven sulfobetainderivaten des tris(2-pyridyl)phosphans als wasserläsliche komplexliganden[J].J Mol Catal,1991,66(2):143-154.
[11] BRAUER D J,SCHENK S,ROSZENBACH S,et al.Water soluble phosphines-Part XIII.Chiral phosphine ligands with amino acid moieties [J].J Organomet Chem,2000,598(1):116-126.
[12] HEINICKE J,PEULECKE N,JONES P G.Novelα-functionally substituted amino acids:diphenylphosphino-glycines [J].Chem Commum,2005,36(21):262-264.
[13] BASKAKOV D,HERRMANN W A.Water-soluble metal complexes and catalysts:Part XI.Novel ligands from tris(hydroxymethyl)phosphaneand amino acids:synthesis and catalytic studiesin two-phase hydroformylation [J].J Mol Catal A:Chem,2008,283:166-170.
[14] EVANS D,OSBORN J A,WILKINSON G J.Hydroformylation of alkenes by use of rhodium complex catalysts[J].J Chem Soc A,1968,9(12):3133-3142.
[15] 尹铎,马昱博,高志贤,等.钴的引入对双环戊二烯氢甲酰化担载Rh催化剂的性能影响[J].化工科技,2014,22(6):1-5.
