抑制COX-2表达通过Akt/iNOS/NO信号通路发挥抗凋亡作用机制的研究
2017-03-04孙小婷麻海春
孙小婷,姜 曦,庞 磊,麻海春*
(吉林大学第一医院1.麻醉科;2.健康管理中心,吉林 长春130021)
抑制COX-2表达通过Akt/iNOS/NO信号通路发挥抗凋亡作用机制的研究
孙小婷1,姜 曦2,庞 磊1,麻海春1*
(吉林大学第一医院1.麻醉科;2.健康管理中心,吉林 长春130021)
目的 探讨心肌缺血/再灌注损伤过程中环氧化酶-2(COX-2)的表达水平与细胞凋亡之间联系及作用机制。方法 通过对心肌细胞进行缺氧/复氧(H/R)实验,模拟心肌缺血再灌注过程,检测COX-2及凋亡相关蛋白的表达水平;采用COX-2特异性抑制剂NS398对其活性进行抑制,观察细胞凋亡水平的变化,并研究其作用机制。结果 与对照组相比,在H/R组中,Bcl2表达明显减少,cleaved capase-3的表达明显增强;在NS398预处理后,cleaved caspase-3的表达明显减少,Bcl2表达增强,TUNEL试验结果提示,NS398预处理明显减少阳性细胞的比例。结论 在心肌细胞中,H/R刺激能够激活COX-2表达,并通过促凋亡途径导致细胞损伤;抑制COX-2活性能够激活Akt/iNOS/NO信号通路,并进而通过抗凋亡机制,发挥对心肌细胞的保护作用。
COX-2;心肌缺血/再灌注损伤;缺氧/复氧;心肌细胞;细胞凋亡
(ChinJLabDiagn,2017,21:0290)
在心肌缺血过程中,细胞中环氧化酶(cyclooxygenase,COX)表达水平显著增高,且心肌梗死时细胞凋亡的严重程度与COX-2表达水平呈正相关,提示COX-2的表达可能与缺血性心肌病相关[1-3]。尽管文献报道在心肌缺血再灌注损伤(MI/ RI)的动物模型中,COX-2的表达具有心肌保护作用,但更多的报告倾向于COX-2表达的增强在MI/ RI中扮演有害角色[4-7]。在大鼠[4-6,8]、兔子[6]和狗[7]等动物模型中,采用COX-2特异性抑制剂对动物进行预处理后,动物的心功能明显提高,心肌梗死面积也有所下降[9]。本研究通过缺氧/复氧(hypoxia/ reoxygenation,H/ R)实验,模拟心肌缺血再灌注过程,分别在心肌细胞株H9C2细胞和原代大鼠心肌细胞中探究COX-2表达水平与Akt/iNOS/NO信号通路之间潜在联系及分子机制。
1 材料和方法
1.1 细胞培养与实验分组
细胞系H9C2购自American Type Culture Collection公司 (ATCC,Manassas,VA,USA),大鼠心肌细胞的分离与培养方法详见文献[10]。分别将H9C2细胞与大鼠心肌细胞分别随机分为三组:对照组(CTL)、缺氧/复氧组(H/R组)和给药组(H/R +NS398组)。前期工作显示,采用10μM COX-2特异性抑制剂NS398对细胞预处理1小时能够明显抑制COX-2的活性。
1.2 MTT试验
培养基中的乳酸盐脱氢酶(LDH)水平是评估细胞损伤的重要指标[11],采用LDH检测试剂盒(Cat.no.11644793001Roche,Mannheim,Germany)分别对H9C2细胞和大鼠心肌细胞的活力进行评估。
1.3 Western blot实验
分别提取各组中H9C2细胞和大鼠心肌细胞中的蛋白,通过western blot对其蛋白表达水平进行检测,所用抗体及其滴度如下:COX-2(1∶1000)、iNOS(1∶40)、GADPH (1∶1000)、总caspase-3 (1∶1000)、 cleaved caspase-3 (1∶1000)、Akt (1∶1000)、p-Akt (1∶1000),各抗体购买公司请参见文献[10]。采用Image J软件对蛋白表达水平进行定量分析。1.4 TUNEL试验
将H9C2细胞接种在载玻片上,在经过不同处理之后,采用原位细胞凋亡检测试剂盒(Cat.no.11684817910,Roche Diagnostics GmbH,Mannheim,Germany)对其进行TUNEL染色,判定心肌细胞凋亡水平。
1.5 COX-2活性检测
采用COX荧光活性分析试剂盒(Cat.no.700200,Cayman)测定不同组别细胞中COX-2的活性。
1.6 测定细胞内一氧化氮(NO)含量
在有或无H/R刺激下,NS398 (10 μM 1小时) 或者 LY294002 (10 μM 1小时)[12]或者1400W (1μM 30分钟)[13]将H9C2细胞培养在96孔黑色的聚苯乙烯板上(Cat.no.3916,Corning Life Sciences)检测细胞内NO。
1.7 统计学分析
所有数据均以平均数±标准差表示,组间比较用Mann-Whitney U测试或者单向方差分析及Tukey’s后检验,使用GraphPad Prism 5.0软件进行做图,P<0.05为有统计学意义。
2 结果
2.1 H/R介导的COX-2表达水平上升
在H/R后,实验组LDH释放明显增多,提示H/R心肌细胞损伤模型构建成功(图1A),且实验组中COX-2的表达显著上升(图1B)。然而,在经过COX-2特异性抑制剂NS398预处理之后,LDH的释放明显减少(图2A),细胞活力明显增强(图2B)。
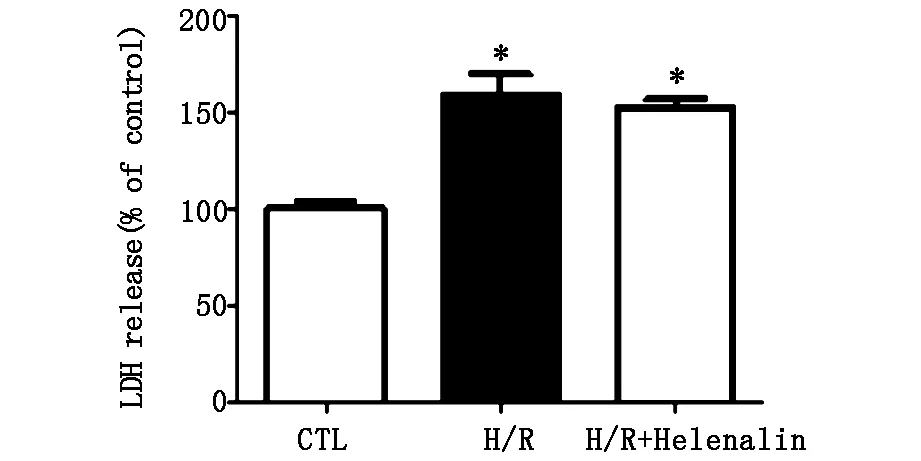
图1A 在H/R后,LDH释放的比较( *P<0.05 CTL vs.H/R or H/R+Helenalin,#P<0.05 H/R vs.H/R+Helenalin)

图1B 在H9C2心肌细胞中,H/R后,COX-2表达的比较( *P<0.05 CTL vs.H/R or H/R+Helenalin,#P<0.05 H/R vs.H/R+Helenalin)

图2A 经过COX-2特异性抑制剂NS398预处理,LDH的释放比较(*P<0.05 CTL vs.H/R or H/R + NS398,#P<0.05 H/R vs.H/R + NS398)

图2B 经过COX-2特异性抑制剂NS398预处理,细胞活力的比较(*P<0.05 CTL vs.H/R or H/R + NS398,#P<0.05 H/R vs.H/R + NS398)
2.2 在H9C2细胞中,抑制COX-2活性能够减轻H/R介导的细胞凋亡
与对照组相比,在H/R组中,Bcl2表达明显减少,cleaved capase-3的表达明显增强。而在NS398预处理后,cleaved caspase-3的表达明显减少,Bcl2表达增强(图3A)。此外,TUNEL试验结果提示,NS398预处理明显减少阳性细胞的比例(图3B)。
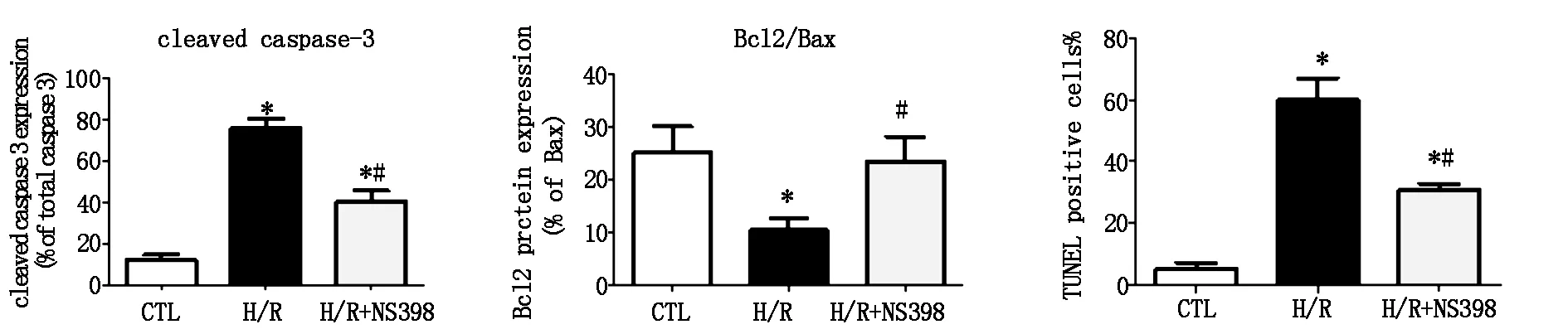
图3A 在H9C2细胞中,cleaved caspase-3、Bcl2表达的比较(*P<0.05 CTL 图3B 在H9C2细胞中,荧光显微镜下阳性
vs.H/R or H/R + NS398,#P<0.05,H/R vs.H/R + NS398) 细胞的比例的比较(*P<0.05,CTLvs.H/R or H/R + NS398;#P<0.05,H/R vs.H/R + NS398)
2.3 成年大鼠心肌细胞中,COX-2参与了H/R介导的细胞损伤与凋亡
H/R明显增加了原代心肌细胞中LDH的释放,cleaved caspase-3的表达,并减弱了细胞活力(图4)。在NS398预处理之后,LDH的释放明显减少(图4A),细胞活力增强(图4B),cleaved caspase-3表达水平降低(图4C)。
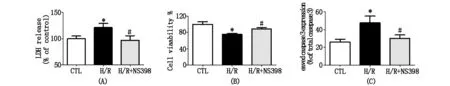
图4 (A,B,C)在原代成年大鼠心肌细胞中,LDH的释放、cleaved caspase-3 表达、细胞活力的比较(*P<0.05 CTL vs.H/R,#P<0.05 H/R vs.H/R + NS398)
2.4 在H9C2细胞中,抑制COX-2活性能够激活Akt/iNOS/NO信号通路
结果显示,NS398预处理后,p-Akt表达水平显著提高(图5A)。在LY294002预处理后,p-Akt的表达受到了明显的抑制(图5A),并失去了对心肌的保护作用,即LDH释放增多、cleaved caspase-3水平上升、细胞活力减弱(图5A,B,C)。同H/R组相比,H/R+NS398组中检测到了高浓度的NO,而这一过程能够被LY294002抑制(图5D)。在H/R+NS398组中,cleaved caspase-3蛋白水平上升,LDH释放增多,细胞活力减弱,细胞内NO水平受到明显抑制。
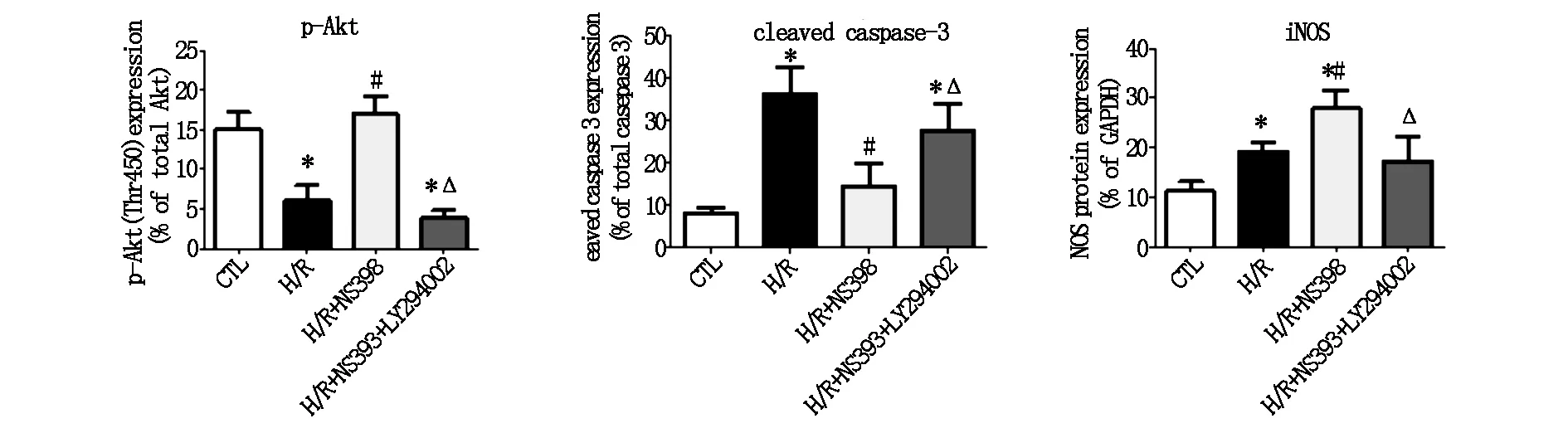
图5(A) 总Akt、p-Akt、cleaved caspase-3、iNOS表达水平的比较(*P<0.05 CTL vs.H/R or H/R + NS398 or H/R+NS398+LY294002,#P<0.05 H/R vs.H/R+NS398,ΔP<0.05 H/R+NS398 vs.H/R+NS398+LY294002)

图5(B) LDH释放水平的比较((*P< 图5(C,D) 细胞活力、NO浓度的比较((*P<0.05 CTLvs.H/R or H/R+0.05 CTL vs.H/R or H/R+ NS398 or H/R+NS398+LY294002,#P<0.05 H/R vs.H/RNS398 or H/R+NS398+LY +NS398,ΔP<0.05 H/R+NS398 vs.H/R+NS398+LY294002)294002,#P<0.05 H/R vs.H/R+NS398,ΔP<0.05 H/R+NS398 vs H/R+NS398+LY294002)
3 讨论
在生理条件下,组织中的COX-2表达水平很低或不表达,前期研究结果显示在不同的心肌缺血再灌注动物模型中,COX-2选择性抑制剂处理可以明显增强心功能,并且减少心肌梗死的面积[4-7]。这一结论与我们在细胞模型实验中的发现一致,然而目前为止,并没有研究阐述抑制COX-2活性是通过何种途径发挥心肌保护作用的。在本研究中,我们通过H/ R实验,模拟心肌缺血再灌注过程,并采用COX-2特异性抑制剂NS398分别作用于H9C2细胞和原始成年大鼠心肌细胞,对其COX-2活性进行特异性抑制,结果显示,抑制COX-2活性能够明显缓解H/R所介导的细胞损伤,如LDH释放量增多、细胞活力下降等。
细胞凋亡主要包括内源性和外源性两种途径[14]。在内源性途径中,当细胞感受到刺激时,促凋亡蛋白(如Bax)破坏线粒体膜结构的完整性,导致线粒体功能障碍和细胞色素的释放,然而抗凋亡蛋白(如Bcl2)则可通过干扰促凋亡细胞聚集来阻止细胞凋亡进程[15,16]。在外源性途径中,配体与细胞表面凋亡受体相结合,形成凋亡复合体并且激活caspase-8和caspase-10,它们能够裂解caspase-3,聚集外源性和内源性半胱天冬酶级联反应[8,14]。研究表明H/R刺激可以通过内源性和外源性途径诱导心肌细胞凋亡[8,17,18],然而心肌细胞受H/R刺激的作用时间不同可能引发不同的凋亡途径。在本研究中,在H/R组中,Bcl2表达明显减少,cleaved capase-3的表达明显增强,这些结果提示H/R刺激可能分别通过内源性和外源性两条途径诱导心肌细胞凋亡。值得注意的是,在H/R刺激下Bax表达水平并没有发生变化,这可能归因于目前实验设置中特异的H/R条件(6小时缺氧和12小时复氧)。采用COX-2特异性抑制剂NS398对细胞进行预处理能明显的减少H/R刺激所介导的上述不良事件,以上结果提示,抑制COX-2活性可能是通过减少细胞凋亡来减轻H/R所介导的细胞损伤。
紧接着我们研究了抑制COX-2是如何减少H/R所介导的细胞凋亡的。Akt信号通路在调控心肌细胞存活与凋亡中扮演重要角色[19]。在本研究中,H/R刺激在诱导COX-2表达的同时伴随着Akt磷酸化水平下降,而抑制COX-2表达则能够促进Akt活化。iNOS是Akt的下游靶点[20],在MI/ IR过程中,活化的Akt能够通过调节iNOS水平,诱导NO合成[19,21]。在本研究中,当H9C2细胞中COX-2表达受到抑制时,细胞内Akt磷酸化水平提高,并伴随着iNOS表达水平和细胞内NO浓度上升,然而,这一过程能够被Akt特异性抑制剂LY294002 或者iNOS 特异性抑制剂1400W 阻断,从而失去对心肌细胞的保护作用。综上,这些观察表明,抑制COX-2可能通过Akt/iNOS/NOS信号通路的活化来防御细胞凋亡。
[1]Schror K,Zimmermann KC,Tannhauser R.Augmented myocardial ischaemia by nicotine--mechanisms and their possible significance[J].Br J Pharmacol,1998,125(1):79.
[2]Wong SC,Fukuchi M,Melnyk P,et al.Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure[J].Circulation,1998,98(2):100.
[3]Abbate A,Santini D,Biondi-Zoccai GG,et al.Cyclo-oxygenase-2 (COX-2) expression at the site of recent myocardial infarction:friend or foe?[J].Heart,2004,90(4):440.
[4]Lada-Moldovan L,Kaloustian S,Bah TM,et al.Chronic pretreatment with celecoxib reduces infarct size[J].J Cardiovasc Pharmacol,2009,54(1):31.
[5]Saito T,Rodger IW,Shennib H,et al.Cyclooxygenase-2 (COX-2) in acute myocardial infarction:cellular expression and use of selective COX-2 inhibitor [J].Can J Physiol Pharmacol,2003,81(2):114.
[6]Saeed SA,Ahmed S.Role of cyclooxygenase-2 in myocardial infarction and ischaemia[J].J Coll Physicians Surg Pak,2006,16(5):324.
[7]Carnieto A,Jr.,Dourado P M,Luz P L,et al.Selective cyclooxygenase-2 inhibition protects against myocardial damage in experimental acute ischemia [J].Clinics (Sao Paulo),2009,64(3):245.
[8]Kunak CS,Kukula O,Mutlu E,et al.The Effect of Etoricoxib on Hepatic Ischemia-Reperfusion Injury in Rats [J].Oxid Med Cell Longev,2015,2015(598162.
[9]Pang L,Cai Y,Tang EH,et al.Prostaglandin E Receptor Subtype 4 Signaling in the Heart:Role in Ischemia/Reperfusion Injury and Cardiac Hypertrophy [J].J Diabetes Res,2016,132(4):347.
[10]Pang L,Ma H,Cai Y,et al.Inhibition Of COX-2/PGE2/EP4 Signaling Protects Against Post-hypoxic Apoptosis In H9C2 Cardiomyocytes [J].The FASEB Journal,2016,30(1 Supplement):939.7.
[11]Legrand C,Bour JM,Jacob C,et al.Lactate dehydrogenase (LDH) activity of the cultured eukaryotic cells as marker of the number of dead cells in the medium [corrected] [J].J Biotechnol,1992,25(3):231.
[12]Liu B,Wang G,Yang J,et al.Berberine inhibits human hepatoma cell invasion without cytotoxicity in healthy hepatocytes [J].PLoS One,2011,6(6):e21416.
[13]Soto M S,O'brien E R,Andreou K,et al.Disruption of tumour-host communication by downregulation of LFA-1 reduces COX-2 and e-NOS expression and inhibits brain metastasis growth [J].Oncotarget,2016,
[14]Elmore S.Apoptosis:a review of programmed cell death [J].Toxicol Pathol,2007,35(4):495.
[15]Burlacu A.Regulation of apoptosis by Bcl-2 family proteins [J].J Cell Mol Med,2003,7(3):249.
[16]Zhang J,Xia Y,Xu Z,et al.Propofol Suppressed Hypoxia/Reoxygenation-Induced Apoptosis in HBVSMC by Regulation of the Expression of Bcl-2,Bax,Caspase3,Kir6.1,and p-JNK [J].Oxid Med Cell Longev,2016,2016(1518738.
[17]Chang G,Zhang D,Liu J,et al.Exenatide protects against hypoxia/reoxygenation-induced apoptosis by improving mitochondrial function in H9c2 cells [J].Exp Biol Med (Maywood),2014,239(4):414.
[18]Li K,Cui Y C,Zhang H,et al.Glutamine Reduces the Apoptosis of H9C2 Cells Treated with High-Glucose and Reperfusion through an Oxidation-Related Mechanism[J].PLoS One,2015,10(7):e0132402.
[19]Matsui T,Tao J,Del Monte F,et al.Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo[J].Circulation,2001,104(3):330.
[20]Oh JH,Lee TJ,Park JW,et al.Withaferin A inhibits iNOS expression and nitric oxide production by Akt inactivation and down-regulating LPS-induced activity of NF-kappaB in RAW 264.7 cells[J].Eur J Pharmacol,2008,599(1-3):11.
[21]Bolli R.Cardioprotective function of inducible nitric oxide synthase and role of nitric oxide in myocardial ischemia and preconditioning:an overview of a decade of research[J].J Mol Cell Cardiol,2001,33(11):1897.
Inhibition of COX-2 protects against cardiomyocyte apoptosis via Akt/iNOS/NO pathway
SUNXiao-ting1,JIANGXi2,PANGLei1,etal.
(1.DepartmentofAnesthesiology,2.HealthManagementCenter,TheFirstHospitalofJilinUniversity,Changchun130021,China)
Objective To explore the potential relationship and mechanism between cyclooxygenase-2 (COX-2) expression and apoptosis during myocardial ischemia /reperfusion injury(MI/ RI).Methods We simulated the process of MI/ RI by hypoxia /reoxygenation (H/R) experiment in both H9C2 cell line and primary rat cardiomyocytes,and detected the expression of COX-2 and apoptosis-related protein.Furthermore,COX-2 expression was inhibited by NS398,COX-2 specific inhibitor.Changes of cell apoptosis were detected,and the underlying mechanism was explored.Results Pre-treatment with NS398 significantly attenuated H/R-induced cell injury and apoptosis [increased expression of Bcl2 and reduced level of cleaved caspases-3 and TUNEL-positive cells] in cardiomyocytes.Conclusion In cardiomyocytes,H/R stimulation will activate COX-2 expression and induce cell damage through apoptotic pathway.Inhibition of COX-2 will protect cardiomyocytes against apoptosis via Akt/iNOS/NO signaling pathway.
COX-2;myocardial ischemia/ reperfusion injury (MI/ RI);hypoxia/reoxygenation (H/R);cardiomyocytes,apoptosis
吉林省教育厅135计划(2014-471);吉林大学研究生创新基金资助项目(2016226);吉林大学第一院第七届青年基金(JDYY72016023)
1007-4287(2017)02-0290-05
R541
A
2016-09-25)
*通讯作者
