药物共晶的合成和结构分析
2017-02-28黄耀辉尹秋响张霞郭明霞王昌
黄耀辉,尹秋响,2,张霞,郭明霞,王昌
(1化学工程联合国家重点实验室,天津大学化工学院,天津 300072;2化学化工协同创新中心,天津 300072)
药物共晶的合成和结构分析
黄耀辉1,尹秋响1,2,张霞1,郭明霞1,王昌1
(1化学工程联合国家重点实验室,天津大学化工学院,天津 300072;2化学化工协同创新中心,天津 300072)
药物的理化性质与其结晶形式相关,药物共晶作为一种新型的固态形式能够在不影响药物内部结构的同时改善药物的多方面性质,提高药效。通过药物共晶的定义、应用、制备方法和结构研究等方面对目前药物共晶的研究现状进行总结,为后续共晶方向的研究提供理论指导。
药物共晶;合成;结晶;结构研究;化学过程
引 言
药物能够以多种不同的固态形式存在,如多晶型、溶剂化合物、盐、共晶和无定形等,每种固态形式都具有自身独特的理化性质,进而影响药物的溶解度、稳定性、生物利用度等性能[1],而药物的疗效很大程度上取决于活性药物成分自身的理化性质及其固体形态。因此,在科学研究和临床医学中确定和选择活性药物成分的最佳固体形态具有重要的作用[2]。
药物共晶作为一种新兴的药物晶型,可以在不影响药物的内部结构的同时改善药物的理化性质,提高临床药效。药物共晶可以增加药物的结晶形式,克服多晶型、盐、溶剂化合物存在的缺点,使其在新药研发及工业生产中具有巨大的应用潜力[3]。
目前药物共晶是改善药物的理化性质、增强药效方面的研究热点。国内外针对药物共晶已有综述发表,主要关于药物共晶的性质改善、设计合成和形成机理等方面。马坤[4]从药物共晶的筛选和溶解行为两方面入手概括总结了共晶的筛选技术和热力学方面的研究进展。王义成等[5]针对活性药物成分的性质和生物利用度等方面分类综述了不同种药物共晶在药学方面的应用。陈学文等[6]系统地介绍了共晶对于原料药理化性质的改善,并对共晶的研发过程进行了总结。高缘等[7]通过介绍药物共晶的形成原理和设计思路等研究进展阐述了共晶在药学领域的应用前景。这些综述都给后续共晶的研究提供了启示和帮助。
在国外方面,Chieng等[8]详尽地介绍了多种药物固态形式的表征分析方法,列举了AMG517、卡马西平和吲哚美辛等多种药物共晶的研究分析手段,对共晶的表征分析方面研究有极大的帮助。Braga等[9]总结了多篇文献,详细地介绍了研磨法在不同种共晶制备方面的应用,证明了研磨制备法在绿色化学方面的优势。Friščić等[10]对研磨法制备共晶给出了全面的阐述,并对研磨法制备共晶的机理做了初步解释,对后续关于共晶形成机理的研究有极大的启发。Thakuria等[11]系统地叙述了药物共晶对活性药物成分溶解度的影响,阐述了形成共晶影响溶解度的机理。Aitipamula等[12]针对共晶的多晶型现象总结概括了共晶多晶型的分类、筛选、伴随现象和理化性质等多方面内容,并列举了大量的共晶多晶型的范例,对共晶的多晶型现象进行了全面的概述。Brittain[13-14]总结了2010年和2011年共晶的研究情况,从研究热点、共晶的制备和表征方法以及包含有药物组分共晶的细节讨论几方面将当年发表在物理学、晶体学和药学期刊上的相关文章进行了系统的概述,对后续共晶的研究有较好的指导意义。
本工作主要从药物共晶的定义、应用、制备方法和结构等方面展开叙述,总结近些年共晶方面的研究进展,为药物共晶的制备和结构的理解分析提供参考。
1 药物共晶的定义和应用
早在19世纪90年代,共晶的典型醌氢醌就已经被文献报道[15]。但共晶这一固态形式被发现以来,其具体定义一直存在争议。目前接受度较高的定义是Salmon等[16]提出的:药物共晶是活性药物成分(active pharmaceutical ingredient,API)分子与其他生理上可接受的酸、碱、盐、非离子化合物分子等共晶形成物(co-crystal former,CCF)以氢键等非共价键相连而结合在同一晶格中。药物共晶组分中至少有一种是分子或离子型的API,同时任意组分在室温下均为固体,并且两种分子有固定的化学计量比。以氢键、π-π堆积作用、范德华力和其他非共价键为基础的分子网状链接方式是共晶形成的基础[17]。
药物共晶的最大优点在于可以在不改变药物共价结构的前提下改变药物的多种理化性质,同时参与形成共晶的配体不同时对药物理化性质的改变方向和程度均不同,因此可以利用制备药物共晶的方法调整活性药物成分的性质,以达到医学以及工业生产上的多种需求。核苷酸类似物抗病毒药物阿德福韦酯,由于能够低剂量治疗乙型肝炎一直在临床上广泛应用,其在人体内水解为阿德福韦发挥抗病毒作用,而阿德福韦由于结构中的多种基团和应用前景,研究人员一直在寻找它的新晶型,Zhang等[18]通过合成3种不同配体的阿德福韦共晶成功地改变了原料药的热稳定性和溶出度等性质,加强了阿德福韦在肠胃的吸收;去铁酮能够有效促进铁的排除,可用于地中海贫血症患者的治疗,但其高溶解度导致的频繁给药限制了临床应用,Zhang等[19]通过合成去铁酮与对羟基苯甲酸、2,5-二羟基苯甲酸共晶和马来酸3种配体的共晶延缓了去铁酮在体内的释放速度,进而改善了药物的高溶解度导致频繁给药的这一缺点,极大地改善了去铁酮的应用效果。
药物共晶还可以改变药物的熔点,增加难溶药物的溶解度,提高活性药物成分的生物利用度,增强药物在储存过程中的稳定性,其他还有降低药物的吸湿性、改变晶型等一系列性质。染料木素较差的水溶性限制了其在多方面的应用,寻找新型的固态形式来提高其溶解度一直以来是这种药物研究的重点,Sowa等[20]将染料木素与咖啡因相结合制备成共晶的形式,使得其在水中的溶解度有大幅度的提高;6-巯基嘌呤是一种重要的临床上用来治疗白血病、系统性红斑狼疮、类风湿性关节炎的药物,其低水溶性使得其口服生物利用度较差(约为16%),而通过与对羟基苯甲酸和2,4-二羟基苯甲酸结合形成共晶,其溶解度分别提升为原来的1.6倍和2倍[21]。McNamara等[22]选用戊二酸作为配体制备了2-[4-(4-氯-2-氟苯氧基)苯基]吡啶-4-甲酰胺的共晶,通过对比原料API和共晶在相同给药量下动物体内的血药浓度证明了形成共晶使得API的生物利用度大幅度提升;阿哌沙班是新型口服抗凝药物,是一种新型口服Xa因子抑制剂,此种药物水溶性较差,口服生物利用度很低,Chen等[23]通过合成阿哌沙班-草酸共晶使其在pH=1的盐酸溶液和pH=6.8的磷酸盐缓冲剂中的溶解度分别提高至原市售晶型的2.2倍和2.1倍,同时生物利用度也有所提高。另外,通过形成共晶,有大量API的熔点、稳定性、溶解速率、吸湿性和力学性能等性质均得到了改善[24-28]。目前,已经有一部分药物共晶应用于临床治疗,如艾司西酞普兰-水合草酸盐[29]、双丙戊酸钠等。
2 药物共晶的制备
药物共晶的制备方法大体可分为溶液合成法和固相研磨法,溶液合成法主要包括冷却结晶、悬浮结晶、缓慢蒸发、溶析结晶等传统的结晶手段,固相研磨法主要包括固体干磨法和溶剂辅助研磨。如图1所示。

图1 药物共晶的制备方法Fig.1 Preparation of pharmaceutical co-crystals
2.1 溶液合成法
在溶液中能够进行共晶的制备合成,是由于形成共晶的活性药物成分(API)能够与配体(CCF)通过形成氢键等非共价键结合形成更为稳定的分子,成为有利的生产趋势。通过溶液法制备共晶的前提是,API与CCF之间的相互作用不仅要强于同种分子之间的相互作用,还要强于单组分与溶剂之间的相互作用,因此选择合适的溶剂是溶液法制备共晶的前提,而在筛选出适合的溶剂后采用适合的溶液结晶方法进行实验也是影响制备过程的重要因素,不同的结晶手段各有利弊[30]。
健美操课的学习评价主要评价学生在课中知识与技能与健美操活动中具体表现,通过评价以提高学生体能与技能水平,健全其人格品质。经过调研发现:在河南省健美操课的学习评价不够系统全面,主要是以技术考核为主,很少学校是对学生的专项身体素质和技能的总体考核,不利于提高学生技能水平和学习能力。
在溶液法合成共晶的过程中,缓慢蒸发和冷却结晶是最为常用的合成手段。Liao等[31]利用乙腈溶液缓慢蒸发得到了吡拉西坦和一类列配体共晶的单晶;Thanigaimani等[32]运用缓慢蒸发方法制备了2-氨基-5-氯吡啶和3-甲基苯甲酸共晶的单晶;Hickey等[33]将卡马西平无水物和糖精晶体添加到甲醇/乙醇混合溶剂中,加热至70℃使其完全溶解,然后冷却至平衡温度30℃,获得了卡马西平-糖精共晶;Remenar等[34]将含有塞来昔布(Cel)和烟酰胺(Nic)的氯仿溶液冷却至室温制备了Cel:Nic=1:1共晶。
缓慢蒸发是通过蒸发含有化学计量比含量的API和配体的溶液得到共晶。其优点在于晶体受蒸发速度影响生长较慢,较易得到单晶;缺点是能耗较大、污染也较大,并且要求两组分在溶液中的溶解度接近,否则易得到共晶和单组分的混合物。冷却结晶与缓慢蒸发相比更为节能,过程较易控制,适用范围更广;但与缓慢蒸发相同,冷却结晶过程中同样有得到共晶和单组分混合产物的风险。在近期的研究中,悬浮结晶和溶析结晶也越来越多地应用于共晶的制备,其中悬浮结晶适用于大量制备共晶产品,Zhang等[35]通过悬浮结晶方法利用氯仿和甲醇的混合溶剂制备得到了茶碱和草酸化学计量比1:2的共晶,Chun等[36]以水作为溶析剂利用甲醇溶液制备得到了高纯度的吲哚美辛-糖精共晶。
2.2 固态研磨法
通过溶液法制备共晶往往受到API和CCF在溶剂中溶解度差异以及溶质与溶剂之间相互作用的限制[37],较为高效的固态研磨法则能够避免产生类似问题,这一方法是通过机械力化学法诱导共晶的生成[38]。Kulla等[39]即通过固态研磨法制备了之前难以在溶液中得到的吡嗪酰胺和草酸的共晶。
固态研磨法分为干磨法和溶剂辅助研磨。干磨法是通过研磨主体药物和配体的混合物,进而得到共晶产品的一种方法[40];溶液辅助研磨是在研磨过程中通过添加少量的溶剂来提高研磨效率的方法[41],溶剂的添加可以有效地提高产物的收率和共晶产品的结晶度[42],并且通过添加溶剂还可以得到干磨法和溶液法无法制备的共晶[43]。Hornedo等[37]在制备咖啡因和柠檬酸共晶的过程中发现,只有在研磨中添加溶剂才能得到预期的产品,而通过干磨API和CCF无法生成共晶。
研磨法被认为是最有效的制备共晶的方法之一,相比耗时较长的溶液合成法方便快捷,可以有效避免单组分溶剂化物的生成,消耗溶剂量很少,符合绿色化学原则,并且更容易控制生成共晶的药物成分和配体的化学计量比。溶液辅助研磨还可用于共晶多晶型的筛选,通过添加极性不同的溶剂生成的共晶可能具有不同的晶型[44]。Li等[45]在使用溶剂辅助研磨制备5-氟尿嘧啶和对羟基苯甲酸共晶时,通过添加水和四氢呋喃这两种极性不同的溶剂分别得到了目标共晶产物的晶型1和晶型2(图2为两种晶型不同的氢键连接方式)。但是,研磨法也有无法纯化产品、不能控制晶体粒径及形态和较难放大生产的缺点。
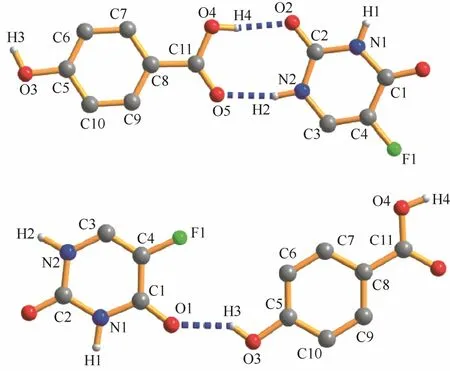
图2 5-氟尿嘧啶和对羟基苯甲酸共晶两种晶型的氢键连接方式[45]Fig.2 Hydrogen bond connection of two forms of 1:1 co-crystal which consist of 5-fluorouracil and 4-hydroxybenzoic acid[45]
此外,除了干磨法和溶剂辅助研磨,也可以将研磨法与其他技术联用来制备共晶。Fulias等[46]先向茶碱和乙酰水杨酸的混合物中添加乙醇进行研磨,然后向研磨产品中再次加入少量乙醇并将其置于微波辐射环境中反应6 min,随后将得到的白色粉末在60℃下静置1 h,干燥得到了纯净的茶碱和乙酰水杨酸共晶。为了避免在溶剂辅助研磨中生成溶剂化物等非期望产品,Hasa等[47]向CAFF-CA、PHE-MA和CAFF-AA 3个体系中添加聚合物PEG辅助研磨,对比干磨法、溶剂辅助研磨法和聚合物辅助研磨法所得产物的XRD、DSC谱图以及通过电子扫描显微镜观察得到的共晶晶体的粒度,得出聚合物可以促进共晶的生成且聚合物的分子量影响所得共晶产品粒度的结论,这一方法开拓了共晶筛选的思路,但添加的聚合物无法分离,因此不能进行实验放大。
2.3 其他制备方法

图3 超临界流体制备共晶流程[49]Fig.3 Schematic description of cocrystallization with supercritical solvent (CSS) setup[49]
3 药物共晶的表征分析方法
共晶可用的表征手段与一般晶体学所用方法类似,单晶X射线分析技术、粉末X射线衍射法(XRD)、热分析法(DSC)和固体红外光谱法等固态表征方法均可用于共晶的性质表征。其中单晶衍射是确定共晶结构的最权威的表征方法[53-54],可以通过单晶衍射得到共晶的晶体结构,但由于较多种类的共晶的单晶难以得到,还需其他固态表征方法辅助研究;粉末X射线衍射则由于其“指纹性”,多数情况下配合DSC曲线一同验证是否生成了共晶;当组分之间形成氢键或卤键等非共价键时,红外光谱的谱图上的特征峰会发生偏移,因此可以根据峰的偏移情况确定是否形成了共晶。
除了对固态产品的表征分析外,许多在线手段和分析方法的联用被运用于共晶形成过程的检测。Lee等[55]通过在线拉曼光谱研究了乙酰水杨酸与4,4′-联吡啶共晶的结晶过程,监测溶液中晶体的成核、生长;Lee等[56]还将拉曼光谱与在线红外光谱联用来研究共晶的成核动力学,图4为其实验装置;Soares等[57]采用拉曼光谱技术对布洛芬和烟酰胺共晶的结晶过程进行记录,通过分析谱图数据得到了生成共晶的终点位置。Lin等[58-59]将DSC与红外光谱联用测定三维的吲哚美辛和糖精共晶的红外谱图(维度上增加了温度),研究了在形成共晶的过程中热量的影响。Sarraguça等[60]运用近红外光谱在线监测呋塞米和对氨基苯甲酸蒸发溶剂形成共晶过程,探究了生成共晶过程中温度的微量差异对共结晶产品的影响。

图4 近红外光谱仪和拉曼光谱联用实验装置[56]Fig.4 Schematic diagram of experimental setup consisting ofin situNIR and Raman spectroscopic analyzers[56]
4 药物共晶的结构
共晶的晶格中包含两种或两种以上的分子,与普通的药物晶体相比包含的分子间作用力种类更多,如氢键、卤键、范德华力、π-π相互作用等,结构更为复杂,从大体上看,基本上是API和CCF先通过氢键、卤键或其他非共价键连接形成相应的带状分子链或片状分子层,然后带与带之间或层与层之间通过弱氢键、范德华力或π-π相互作用连接形成稳定的晶体。具体形式可分为以下几种。
4.1 通过π-π堆积作用直接连接
通常API分子和CCF通过氢键形成简单的二聚体或多聚体,不同的多聚体之间通过范德华力或π-π堆积作用连接组成三维晶体的结构,是最为简单的共晶结构。但由于形成共晶的分子结构通常具有多个官能团和氢键供受体,与周围分子形成非共价键的概率很高,因此这种较为简单的结构并不常见。
Heiden等[61]通过茶碱(TP)和苯甲酸(BA)共晶的二维的X射线衍射谱图,借助模拟手段计算得出茶碱-苯甲酸共晶的晶体结构。在共晶晶体中,茶碱分子和苯甲酸分子通过两条强氢键连接形成二聚体,组成化学计量比为1:1的共晶,其中一条氢键存在于茶碱的羰基与苯甲酸的羧基之间(O—H…O, 0.26149 nm, 168°),另一条氢键形成于茶碱分子咪唑环上的N原子与苯甲酸分子中羧基的双键氧之间(N—H…O, 0.27630 nm, 169°)。茶碱与苯甲酸形成的不同二聚体之间平行排布,平面间距约为0.3551 nm,而通常两个环状结构之间的垂直距离在0.33~0.38 nm之间时能够形成π-π堆积作用[62],因此在茶碱-苯甲酸共晶结构中平行的二聚体之间通过苯甲酸的苯环与茶碱的六元环之间的π-π堆积作用形成三维结构,如图5所示。
4.2 分子链之间连接构建三维结构
在部分共晶结构中,API与CCF以氢键连接形成分子链,然后分子链之间再以非共价键相连组成三维晶体结构。在阿比朵尔(Arb)和琥珀酸(Suc)形成的共晶结构中,Arb和Suc先以1:1的化学计量比通过氢键形成带状结构,然后相邻的分子带通过π-π相互作用和范德华力作用结合形成晶体[63],其结构如图6所示。
4.3 分子链连接成面,面连接成三维结构
API和CCF之间先通过氢键连接形成分子链,然后分子链再依靠氢键或范德华力连接进一步形成二维层状结构,最后层与层之间通过π-π堆积作用形成三维结构,这种模式在共晶的结构中较为常见,大部分共晶以这种方式形成三维立体结构。Krishna等[64]在研究6-氯-2,4-二硝基苯胺(cda)与香兰素(van)的几种同分异构体形成的共晶时,得到的van与cda形成的共晶结构为:一个cda分子与两个van分子以首尾相连的方式通过O—H…O [0.2632(2) nm, 143°]和N—H…O [0.2878(3) nm, 137°]两条氢键连接先形成一维带状结构,不同的分子带之间通过范德华力和C—H…O [0.3499(3) nm, 172°]作用形成二维网状结构,不同的平面网络进一步以π-π堆积作用相结合。具体结构如图7所示。
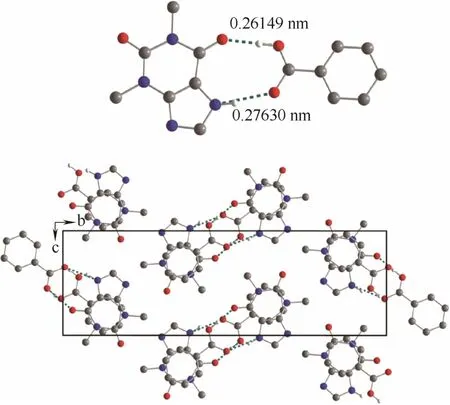
图5 茶碱和苯甲酸形成二聚体结构以及茶碱-苯甲酸共晶的层状结构[61]Fig. 5 Dimer of TP and BA and layered co-crystal structure of TP:BA[61]

图6 阿比朵尔-琥珀酸共晶的分子氢键连接结构(a)和带状结构连接方式(b)[63]Fig.6 Hydrogen-bonded molecular unit in Arb and Suc co-crystal(a), and molecular packing arrangement of Arb and Suc co-crystal(b)[63]
吲哚美辛(IND)和糖精(SAC)在构成共晶时,先分别组成吲哚美辛二聚体和糖精二聚体交互分布,相邻的二聚体垂直排列以减小位阻效应,不同的二聚体之间通过N—H…O [N…O;0.3180 (3) nm, N—H…O;117.18°]氢键连接成链状结构,吲哚美辛二聚体和糖精二聚体分子链之间通过C—H…Cl连接成二维平面,整体结构通过C—H…O氢键稳定[65]。
4.4 螺旋形结构相连接
Sanphui等[66]对比伏立康唑(VOR)与几种有机酸分别形成的盐类和共晶的晶体结构发现,伏立康唑与富马酸(FUM)、对羟基苯甲酸(PHB)和对氨基苯甲酸(PAB)形成的3种共晶的晶体结构类似(图8),为3种有机酸上的羧基、羟基与氨基基团同时与伏立康唑分子结构中的嘧啶环和三唑环上的N原子形成氢键,然后组成一种螺旋结构,每个螺旋结构通过较弱的C—H…F键相连,形成分子网络(图9)。
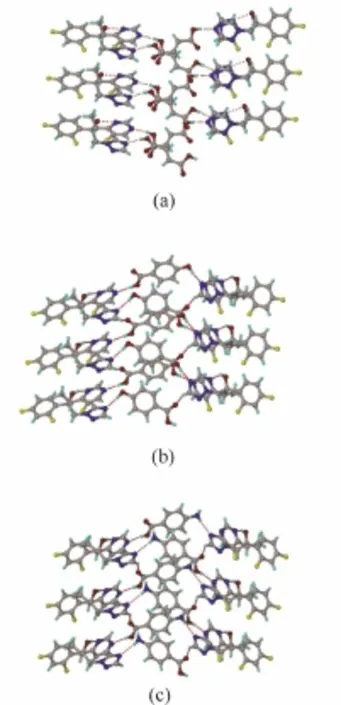
图8 VOR-FUM(a)、VOR-PHB(b)和VOR-PAB(c)3种共晶的分子排布和氢键网络[66]Fig. 8 Molecular packing and hydrogen bonded networks in crystal structure of VOR-FUM (a), VOR-PHB (b), and VOR-PAB(c) co-crystals[66]
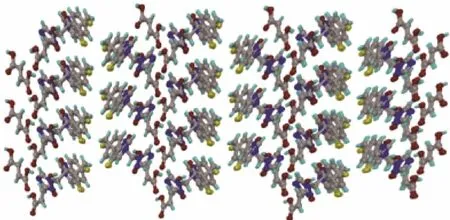
图9 VOR-FUM共晶的三维分子排布和氢键网络[66]Fig. 9 Three-dimensional molecular arrangement and hydrogen bond network of VOR-FUM co-crystal[66]
5 焦点与展望
药物共晶在多个方面表现出相对于传统晶型的性能优势,因此近年来逐渐受到研究人员的广泛关注。目前共晶领域的研究热点主要集中于以下几个方面。
(1)新型共晶的筛选和制备。自共晶这一物质形式被发现以来,各种新型共晶的筛选便一直是研究人员研究的主攻方向。目前药物研究领域的许多API在溶解度和生物利用度等方面均面临一定程度的限制,需要以频繁给药或加大用药剂量等方式来确保疗效,研究人员一直致力于寻找合适的固体形式来克服其临床应用的缺点[67-69],而共晶这一固态形式能够在多个方面满足对药物改性的需求,因此新型共晶的筛选和制备在未来一段时间仍然会是人们的研究热点。
(2)新型制备方法的推广。近年来,超临界流体法、超声法、熔融法和微波法等新型制备方法的应用日渐广泛[70],一部分通过传统方法无法得到共晶的药物通过这些新型方法筛选出了新共晶,并且新型方法通常更符合绿色化学的要求,因此更加受到人们的青睐,而简化实验装置、降低成本、运用新型制备方法提高共晶的筛选效率也成为目前共晶领域的一大研究焦点。
(3)共晶合成过程研究。相比过去10年专注于药物共晶本身和物化性质的改善,通过在线拉曼光谱、在线红外光谱等手段对合成共晶的过程在线监控成为大多数研究人员的主要研究内容[71],由于影响共晶形成的因素还有许多是未知的,对共晶的合成过程的研究就尤为重要,而对这一过程的研究也将推动对共晶形成机理的进一步的深入理解,因此是共晶目前的研究热点中最为重要的一部分。
目前人们对于药物共晶对原料药性质的改善已经有了较为深入的认识,相比其他固态形式的优势也有了一定程度的了解,但对于影响共晶形成的关键性因素和共晶的形成机理还研究得不够全面,所以虽然结合晶体工程学和超分子化学来设计制备药物共晶初有成效,却仍然无法准确预测哪些药物和配体能够形成共晶,在药物共晶的设计合成过程中前期大量的筛选实验和较低的成功率依然无法避免。因此,对共晶形成机理的进一步探究是未来共晶领域的重点研究方向,如溶液合成法中不同溶剂所起的作用、固态研磨法中API和配体如何发生作用相结合、超声和微波等手段如何促进药物和配体之间通过非共价键连接,这些问题都有待研究人员去探究解决。
6 结 论
随着对共晶问题研究的深入,共晶的概念逐渐进入人们的视野,人们发现共晶在改善药物物理化学性质方面有很好的作用,可以提高API的溶解度,增加稳定性、生物利用度和溶出速率等,而且共晶稳定性好,共晶的制备过程既省时又省钱,因此作为一种新的药物制剂方式,共晶及其制备方法近年来得到了药物研究界的广泛关注。目前已有部分共晶药物应用于临床应用,未来共晶在工业和医疗产业中必将发挥更广阔的作用。
[1] BYRN S R, ZOGRAFI G, CHEN X M. Accelerating proof of concept for small molecule drugs using solid-state chemistry[J]. Journal of Pharmaceutical Sciences, 2010, 99(9): 3665-3675.
[2] SCHULTHEISS N, NEWMAN A. Pharmaceutical cocrystals and their physicochemical properties[J]. Crystal Growth & Design, 2009, 9(6): 2950-2967.
[3] MORISSETTE S L, ALMARSSON O, PETERSON M L,et al. High-throughput crystallization: polymorphs, salts, co-crystals and solvates of pharmaceutical solids[J]. Advanced Drug Delivery Reviews, 2004, 56(3): 275-300.
[4] 马坤. 药物共晶的筛选技术及热力学研究进展[J]. 药学进展, 2010, 34(12): 529-534. MA K. Progress in the research of screening technique and thermodynamics of pharmaceutical cocrystals[J]. Progress in Pharmaceutical Sciences, 2010, 34(12): 529-534.
[5] 王义成, 冯成亮, 杨素勤, 等. 药物共晶的最新研究进展[J]. 药学进展, 2013, 37(3): 120-130. WANG Y C, FENG C L, YANG S Q,et al. Recent research advances of pharmaceutical cocrystals[J]. Progress in Pharmaceutical Sciences, 2013, 37(3): 120-130.
[6] 陈学文, 宋菊, 唐海谊, 等. 药物共晶筛选与理化性质研究进展[J].中国医药工业杂志, 2012, 43(8): 703-708. CHEN X W, SONG J, TANG H Y,et al. Progress in screening methods and physicochemical properties of pharmaceutical co-crystals[J]. Chinese Journal of Pharmaceuticals, 2012, 43(8): 703-708.
[7] 高缘, 祖卉, 张建军. 药物共晶研究进展[J]. 化学进展, 2010, 22(5): 829-836. GAO Y, ZU H, ZHANG J J. Pharmaceutical cocrystals[J]. Progress in Chemistry, 2010, 22(5): 829-836.
[8] CHIENG N, RADES T, AALTONEN J. An overview of recent studies on the analysis of pharmaceutical polymorphs[J]. Journal of Pharmaceutical and Biomedical Analysis, 2011, 55(4): 618-644.
[9] BRAGA D, MAINI L, GREPIONI F. Mechanochemical preparation of co-crystals[J]. Chemical Society Reviews, 2013, 42(18): 7638-7648.
[10] FRIŠČIĆ T, JONES W. Recent advances in understanding the mechanism of cocrystal formationviagrinding[J]. Crystal Growth & Design, 2009, 9(3): 1621-1637.
[11] THAKURIA R, DELORI A, JONES W,et al. Pharmaceutical cocrystals and poorly soluble drugs[J]. International Journal of Pharmaceutics, 2013, 453(1): 101-125.
[12] AITIPAMULA S, CHOW P S, TAN R B H. Polymorphism in cocrystals: a review and assessment of its significance[J]. Cryst. Eng. Comm., 2014, 16(17): 3451-3465.
[13] BRITTAIN H G. Cocrystal systems of pharmaceutical interest: 2010[J]. Crystal Growth & Design, 2012, 12(2): 1046-1054.
[14] BRITTAIN H G. Cocrystal systems of pharmaceutical interest: 2011[J]. Crystal Growth & Design, 2012, 12(11): 5823-5832.
[15] LING A R, BAKER J L. Halogen derivatives of quinine(Ⅲ): Derivatives of quinhydrone[J]. J. Chem. Soc. Trans., 1893, 63: 1314-1327.
[16] AAKEROY C B, SALMON D J. Building co-crystals with molecular sense and supermolecular sensibility[J]. Cryst. Eng. Comm., 2005, 7: 439-448.
[17] RODRIGUEZ H N. Cocrystals: molecular design of pharmaceutical materials[J]. Molecular Pharmaceutics, 2007, 4(3): 299-300.
[18] ZHANG X, SUN F, ZHANG T,et al. Three pharmaceuticals cocrystals of adefovir: syntheses, structures and dissolution study[J]. Journal of Molecular Structure, 2015, 1100: 395-400.
[19] ZHANG X, TIAN Y, JIA J,et al. Synthesis, characterization and dissolution of three pharmaceutical cocrystals based on deferiprone[J]. Journal of Molecular Structure, 2016, 1108: 560-566.
[20] SOWA M, SLEPOKURA K, MATCZAL-JON E. Solid-state characterization and solubility of a genistein-caffeine cocrystal[J]. Journal of Molecular Structure, 2014, 1076: 80-88.
[21] XU L L, CHEN J M, YAN Y,et al. Improving the solubility of 6‑mercaptopurineviacocrystals and salts[J]. Crystal Growth & Design, 2012, 12(12): 6004-6011.
[22] MCNAMARA D P, CHILDS S L, GIORDANO J,et al. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API[J]. Pharmaceutical Research, 2006, 23(8): 1888-1897.
[23] CHEN Y, LI L, YAO J,et al. Improving the solubility and bioavailability of apixabanviaapixaban-oxalic acid cocrystal[J]. Crystal Growth & Design, 2016, 16(5): 2923-2930.
[24] SHAYANFAR A, ZEYNALI K A, JOUYBAN A. Solubility and dissolution rate of a carbamazepine-cinnamic acid cocrystal[J]. Journal of Molecular Liquids, 2013, 187: 171-176.
[25] DUGGIRALA N K, SMITH A J, WOJTAS L. Physical stability enhancement and pharmacokinetics of a lithium ionic cocrystal with glucose[J]. Crystal Growth & Design, 2014, 14(11): 6135-6142.
[26] WANG L, WEN X N, LI P,et al. 2:1 5-Fluorocytosine-acesulfame CAB cocrystal and 1:1 5-fluorocytosine-acesulfame salt hydrate with enhanced stability against hydration[J]. Cryst. Eng. Comm., 2014, 16(36): 8537-8545.
[27] IMCHALEE R, CHAROENCHAITRAKOOL M. Gas anti-solvent processing of a new sulfamethoxazole-L-malic acid cocrystal[J]. Journal of Industrial and Engineering Chemistry, 2015, 25: 12-15.
[28] ZHOU Z, LI W, SUN W,et al. Resveratrol cocrystals with enhanced solubility and tabletability[J]. International Journal of Pharmaceutics, 2016, 509(1/2): 391-399.
[29] FRAMPTON C. Cocrystal clear solutions[J]. Chemistry & Industry, 2010, (5): 21-23
[30] AAKEROY C B, SALMON D J, SMITH M M,et al. Cyanophenyloximes: reliable and versatile tools for hydrogen-bond directed supramolecular synthesis of cocrystals[J]. Crystal Growth & Design, 2006, 6(4): 1033-1042.
[31] LIAO X, GAUTAM M, GRILL A,et al. Effect of position isomerism on the formation and physicochemical properties of pharmaceutical co-crystals[J]. Journal of Pharmaceutical Sciences, 2010, 99(1): 246-254.
[32] THANIGAIMANI K, KHALIB N C, TEMEL E,et al. Newsupramolecular cocrystal of 2-amino-5-chloropyridine with 3-methylbenzoic acids: syntheses, structural characterization, hirshfeld surfaces and quantum chemical investigations[J]. Journal of Molecular Structure, 2015, 1099: 246-256.
[33] HICKEY M B, PETERSON M L, SCOPPETTUOLO L A,et al. Performance comparison of a co-crystal of carbamazepine with marketed product[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2007, 67(1): 112-119.
[34] REMENAR J F, PERTERSON M L, STEPHENS P W,et al. Celecoxib: nicotinamide dissociation: using excipients to capture the cocrystal's potential[J]. Molecular Pharmaceutics, 2007, 4(3): 386-400.
[35] ZHANG S, RASMUSON. The theophylline-oxalic acid co-crystal system: solid phases, thermodynamics and crystallization[J]. Cryst. Eng. Comm., 2012, 14(14): 4644-4655.
[36] CHUN N H, WANG I C, LEE M J,et al. Characteristics of indomethacin-saccharin (IMC-SAC) co-crystals prepared by an anti-solvent crystallization process[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2013, 85(3): 854-861.
[37] HORNEDO N R, NEHM S J, SEEFELDT K F,et al. Reaction crystallization of pharmaceutical molecular complexes[J]. Molecular Pharmaceutics, 2006, 3(3): 362-367.
[38] KARKI S, FRISCIC T, JONES W. Control and interconversion of cocrystal stoichiometry in grinding: stepwise mechanism for the formation of a hydrogen-bonded cocrystal[J]. Cryst. Eng. Comm., 2009, 11(3): 470-481.
[39] KULLA H, GREISER S, BENEMANN S,et al.In situinvestigation of a self-accelerated cocrystal formation by grinding pyrazinamide with oxalic acid[J]. Molecules, 2016, 21(7): 917-925.
[40] YAMAMOTO K, TSUTSUMI S, LKEDA Y. Establishment of cocrystal cocktail grinding method for rational screening of pharmaceutical cocrystals[J]. International Journal of Pharmaceutics, 2012, 437(1/2): 162-171.
[41] LIN H L, WU T K, LIN S Y. Screening and characterization of cocrystal formation of metaxalone with short-chain dicarboxylic acids induced by solvent-assisted grinding approach[J]. Thermochimica Acta, 2014, 575: 313-321.
[42] NGUYEN K L, FRISCIC T, DAY G M,et al. Terahertz time-domain spectroscopy and the quantitative monitoring of mechanochemical cocrystal formation[J]. Nature Materials, 2007, 6: 206-209.
[43] FRIŠČIĆ T, FABIAN L, BURLEY J C,et al. Exploring the relationship between cocrystal stability and symmetry: is Wallach's rule applicable to multi-component solids?[J]. Chem. Comm., 2008, (14): 1644-1646.
[44] TRASK A V, MOTHERWELL W D, JONES W. Solvent-drop grinding: green polymorph control of cocrystallisation[J]. Chem. Comm., 2004, (7): 890-891.
[45] LI S, CHEN J M, LU T B,et al. Synthon polymorphs of 1:1 co-crystal of 5-fluorouracil and 4-hydroxybenzoic acid: their relative stability and solvent polarity dependence of grinding outcomes[J]. Cryst. Eng. Comm., 2014, 16(28): 6450-6458.
[46] FULIAS A, SOICA C, LEDETI I,et al. Characterization of pharmaceutical acetylsalicylic acid-theophylline cocrystal obtained by slurry method under microwave irradiation[J]. Revista De Chimie, 2014, 65(11): 1281-1284.
[47] HASA D, RAUBER G S, VOINOVICH D,et al. Cocrystal formation through mechanochemistry: from neat and liquid-assisted grinding to polymer-assisted grinding[J]. Angewandte Chemie, 2015, 54(25): 7371-7375.
[48] STAHLY G P. Diversity in single- and multiple-component crystals. The search for and prevalence of polymorphs and cocrystals[J]. Crystal Growth & Design, 2007, 7(6): 1007-1026.
[49] PADRELA L, RODRIGUES M A, TIAGO J,et al. Insight into the mechanisms of cocrystallization of pharmaceuticals in supercritical solvents[J]. Crystal Growth & Design, 2015, 15(7): 3175-3181.
[50] NEUROHR C, ERRIGUIBLE A, LAUGIER S,et al. Challenge of the supercritical antisolvent technique SAS to prepare cocrystal-pure powders of naproxen-nicotinamide[J]. Chemical Engineering Journal, 2016, 303: 238-251.
[51] BERRY D J, SEATON C C, CLEGG W,et al. Applying hot-stage microscopy to co-crystal screening: a study of nicotinamide with seven active pharmaceutical ingredients[J]. Crystal Growth & Design, 2008, 8(5): 1697-1712.
[52] PATIL S, KULKARNI J, MAHADIK K. Exploring the potential of electrospray technology in cocrystal synthesis[J]. Industrial & Engineering Chemistry Research, 2016, 55(30): 8409-8414.
[53] CHILDS S L, STAHLY G P, PARK A. The salt-cocrystal continuum:the influence of crystal structure on ionization state[J]. Molecular Pharmaceutics, 2007, 4(3): 323-338.
[54] TAYLOR R, KENNARD O. Comparison of X-ray and neutron diffraction results for the NH···O=C hydrogen bond[J]. Acta Crystallographica Section B, 1983, 39: 133-138.
[55] LEE K S, KIM K J, ULRICH J.In situmonitoring of cocrystallization of salicylic acid-4,4′-dipyridyl in solution using Raman spectroscopy[J]. Crystal Growth & Design, 2014, 14(6): 2893-2899.
[56] LEE N J, CHUN N H, KIM M J,et al.In situmonitoring of antisolvent cocrystallization by combining near-infrared and Raman spectroscopies[J]. Crystal Growth & Design, 2015, 15(9): 4385-4393.
[57] SOARES F L, CARNEIRO R L. Green synthesis of ibuprofen-nicotinamide cocrystals and in-line evaluation by Raman spectroscopy[J]. Crystal Growth & Design, 2013, 13(4): 1510-1517.
[58] ZHANG G C, LIN H L, LIN S Y. Thermal analysis and FTIR spectral curve-fitting investigation of formation mechanism and stability of indomethacin-saccharin cocrystalsviasolid-state grinding process[J]. Journal of Pharmaceutical and Biomedical Analysis, 2012, 66: 162-169.
[59] LIN H L, ZHANG G C, LIN S Y. Real-time co-crystal screening and formation between indomethacin and saccharinviaDSC analytical technique or DSC-FTIR microspectroscopy[J]. Journal of Thermal Analysis and Calorimetry, 2015, 120(1): 679-687.
[60] SARRAGUÇA M C, PAISANA M, PINTO J,et al. Real-time monitoring of cocrystallization processes by solvent evaporation: a near infrared study[J]. European Journal of Pharmaceutical Sciences, 2016, 90: 76-84.
[61] HEIDEN S, TROBS L, WENZEL K J,et al. Mechanochemical synthesis and structural characterisation of a theophylline-benzoic acid cocrystal (1:1)[J]. Cryst. Eng. Comm., 2012, 14(16): 5128-5129.
[62] JANIAK C. A critical account on π-π stacking in metal complexes with aromatic nitrogen-containing ligands[J]. Journal of the Chemical Society-Dalton Transactions, 2000, (21): 3885-3896.
[63] MANIN A N, SUROV A O, CHURAKOV A V,et al. Crystalstructures, thermal analysis, and dissolution behavior of new solid forms of the antiviral drug arbidol with dicarboxylic acids[J]. Crystals, 2015, 5(4): 650-669.
[64] KRISHNA G R, SHI L, BAG P P,et al. Correlation among crystal structure, mechanical behavior, and tabletability in the co-crystals of vanillin isomers[J]. Crystal Growth & Design, 2015, 15(4): 1827-1832
[65] BASAVOJU S, BOSTROM D, VELAGA S P. Indomethacinsaccharin cocrystal: design, synthesis and preliminary pharmaceutical characterization[J]. Pharmaceutical Research, 2008, 25(3): 530-541.
[66] SANPHUI P, MISHRA M K, RAMAMURTY U,et al. Tuning mechanical properties of pharmaceutical crystals with multicomponent crystals: voriconazole as a case study[J]. Molecular Pharmaceutics, 2015, 12(3): 889-897.
[67] HIENDRAWAN S, VERIANSYAH B, WIDJOJOKUSUMO E,et al. Physicochemical and mechanical properties of paracetamol cocrystal with 5-nitroisophthalic acid[J]. International Journal of Pharmaceutics, 2016, 497(1/2): 106-113.
[68] STAVROPOULOS K, JOHNSTON S C, ZHANG Y,et al. Cocrystalline solids of telaprevir with enhanced oral absorption[J]. Journal of Pharmaceutical Sciences, 2016, 104(10): 3343-3350.
[69] SHETE A, MURTHY S, KORPALE S,et al. Cocrystals of itraconazole with amino acids: screening, synthesis, solid state characterization,in vitrodrug release and antifungal activity[J]. Journal of Drug Delivery Science and Technology, 2015, 28: 46-55.
[70] YAN Y, CHEN J M, LU T B. Thermodynamics and preliminary pharmaceutical characterization of a melatonin-pimelic acid cocrystal prepared by a melt crystallization method[J]. Cryst. Eng. Comm., 2015, 17(3): 612-620.
[71] DING P, LIN L, LI Y,et al.In-situsynchrotron wide-angle X-ray diffraction as a rapid method for cocrystal/salt screening[J]. International Journal of Pharmaceutics, 2015, 496(1): 107-116.
Synthesis and structural analysis of pharmaceutical co-crystals
HUANG Yaohui1, YIN Qiuxiang1,2, ZHANG Xia1, GUO Mingxia1,WANG Chang1
(1State Key Laboratory of Chemical Engineering,School of Chemical Engineering and Technology,Tianjin University,Tianjin300072,China;2Collaborative Innovation Center of Chemical Science and Chemical Engineering,Tianjin300072,China)
Co-crystals have advantages in physiochemical properties over their constituent components and are expected to use in product formulation such as enhanced active pharmaceutical ingredients, food components with improved absorbability, and specialty chemicals with better performance. The development of pharmaceutical co-crystals might offer advantages over the active pharmaceutical ingredients and overcome some of the limitations encountered with classical strategy (polymorph, solvate and salt formation). In recent years, co-crystals have recently gained much attention for pharmaceutical development especially because it has great advantages in improving the solubility, dissolution, melt point and oral bioavailability. Since the co-crystals lattice comprises two or more kinds of molecules, compared with the conventional structure of the drug crystal, intermolecular force comprising more types such as hydrogen, halogen bond, van der Waals forces, π-π interaction, so the structure is more complex. The research on the co-crystals structure can be helpful to understand the formation mechanism. The definition, application, preparation and structure of co-crystals were reviewed, it will provide theoretical guidance for the studies of the following research.
pharmaceutical co-crystals; synthesis; crystallization; structural study; chemical processes
YIN Qiuxiang, qxyin@tju.edu.cn
TQ 460.1
:A
:0438—1157(2017)02—0509—10
10.11949/j.issn.0438-1157.20160928
2016-06-30收到初稿,2016-10-20收到修改稿。
联系人:尹秋响。
:黄耀辉(1993—),女,博士研究生。
天津市应用基础及前沿技术研究计划项目(天津市自然科学基金重点项目)(11JCZDJC20700)。
Received date: 2016-06-30.
Foundation item: supported by the Tianjin Research Program of Application Foundation and Advanced Technology (the Key Program of Natural Science Foundation of Tianjin) (11JCZDJC20700).
