吡咯亚胺钒络合物催化制备含功能性乙氧基硅烷聚乙烯
2017-02-28刘智颖范宏
刘智颖,范宏
(化学工程联合国家重点实验室,浙江大学化学工程与生物工程学院,浙江 杭州 310027)
吡咯亚胺钒络合物催化制备含功能性乙氧基硅烷聚乙烯
刘智颖,范宏
(化学工程联合国家重点实验室,浙江大学化学工程与生物工程学院,浙江 杭州 310027)
通过二甲基乙氧基硅烷与1,7-辛二烯的硅氢加成反应,制备出一种含功能性基团的极性单体——7-辛烯基乙氧基二甲基硅烷(OEMS),在吡咯亚胺钒络合物(VCIP)的催化作用下,该单体可与乙烯配位共聚制备含功能性乙氧基硅烷的乙烯共聚物(PE-co-OEMS)。通过高温核磁共振氢谱及碳谱(1H NMR和13C NMR)、高温凝胶渗透色谱(HT-GPC)和热分析(DSC)等测试方法对共聚物的结构和热性能进行了表征。结果显示,当共单体OEMS的投料量增大至60 mmol·L-1时,共聚反应活性仍可保持在1.2 kg·mmol-1·h-1,OEMS单体插入率为1.3%(摩尔分数);此外,OEMS投料量对于共聚物的分子量(Mw)和分子量分布(PDI)均具有较大的影响,当OEMS投料量为30 mmol·L-1时,共聚物Mw为16900,PDI为2.1;若继续增大共单体的投料量,共聚物分子量增大且分子量分布变宽。熔点(Tm)和结晶焓(ΔHc)随着OEMS插入率的增大而降低。在三氟甲磺酸作用下,PE-co-OEMS交联后凝胶含量接近100%。
聚乙烯;有机硅;交联;聚合物;制备;合成
引 言
聚乙烯(PE)材料基于质量轻、成本低和可替代性强等优点,在现代工业和社会生活中应用极为广泛[1]。然而,由于分子中缺乏功能性基团,聚乙烯的黏结性和染色性较差,限制其在材料助剂和黏结等领域的应用范围[2-3]。近来,人们开始越来越多地关注聚烯烃功能化改性的相关研究[4-10]。根据实现方式的不同,聚烯烃的改性分为前功能化改性和后功能化改性。前功能化改性指在聚烯烃合成过程中引入反应性共聚单体;后功能化改性指直接通过化学或物理方法在聚烯烃上引入功能性基团。其中,通过烯烃与功能单体发生共聚反应,是最直接有效的改性方式。然而,由于绝大部分功能性单体对过渡金属催化剂有一定毒化作用,直接共聚法往往难以实现[11]。
为了确保直接共聚法成功引入功能性基团,需要选择合适的催化体系。自Ziegler-Natta催化剂问世以来,钒系催化体系不仅在高分子量聚烯烃、乙丙橡胶等领域获得广泛应用[12-15],部分结构新颖的钒系催化剂还可以用于乙烯和其他功能性单体共聚,所制备的聚合物展现出活性高、分子量分布窄等优势[16-18]。2009年,Xu等[19]合成了一系列吡咯亚胺钒(Ⅲ)配合物,在Et2AlCl和三氯乙酸乙酯的活化作用下,可高效催化乙烯与十一烯醇共聚,插入率达到15.8%时,反应活性仍可保持在1.4 kg·mmol-1·h-1·bar-1(1 bar=0.1 MPa),分子量分布低于2.0,显示出该吡咯亚胺型钒系催化剂在乙烯与极性功能性单体共聚方面具有一定的应用潜力。
有机硅材料通常含有键能较高的Si—O键,其中Si原子与其他有机基团相连,特殊的结构赋予其优良的性能[20],例如可交联性、低表面能、耐候性、热稳定性等,但其力学性能较差,与大部分有机物不能相容[21-23]。因此,若能将有机硅化合物与聚乙烯相结合,合成兼具两者优势的产物,将具有重要的研究意义[24-26]。目前,已有相关文献报道可通过硅氢加成反应[27-28]、接枝共聚[25,29]等多种化学方法将聚烯烃链段与有机硅链段相结合,其中配位共聚法合成聚烯烃有机硅接枝共聚物具有良好的结构调控性,通常在较为温和的常压反应条件下,利用适当的催化剂即可实现烯烃与有机硅极性单体的高效共聚,由此以化学方法实现对聚烯烃材料的功能化改性。2004年,Ciolino等[29]用Et(Ind)2ZrCl2/MAO共聚乙烯与含端乙烯基的聚硅氧烷(PDMS)大单体,制备出PE-PDMS接枝共聚物。Jin等[30]在VCl3(THF)3/Et2AlCl/三氯乙酸乙酯的催化体系下,通过乙烯与1-辛烯-3-辛烷-1,1,3,3-四甲基二硅氧烷(OO7)共聚,合成了新型含硅氧烷的乙烯共聚物。该共聚物重均分子量约为13000,PDI约为2,共单体插入率高达4.9%(摩尔分数)。
烷氧基硅烷功能化的聚烯烃材料因其烷氧基团可进行缩合交联的性质获得了较为广泛的商业应用,交联后的聚烯烃材料具有更加优异的力学性能、化学性能、抗应力裂变性能和热性能,除此之外,烷氧基硅烷的缩合交联反应可简单便捷地实现交联材料的制备。2013年,Zimmer等[31]通过将两步反应制得二叔丁氧基甲基辛烯基硅氧烷,并成功将其与丙烯共聚制得一种新型的交联共聚物,该共聚物在三氟甲磺酸作用下交联后,凝胶含量可达到98%。然而,该种有机硅单体所含的叔丁氧基因位阻较大导致交联反应活性较弱,且在单体浓度达到33 mmol·L-1时,未经交联的共聚产物会产生凝胶,共聚反应的活性也较低;如果能够使用更加高效的催化剂,实现烯烃与交联反应活性更高的烷氧基硅烷共聚,将具有更实际的应用意义。
本文合成了一种结构清晰且交联反应性较强的有机硅极性单体7-辛烯基乙氧基二甲基硅烷(OEMS),在吡咯亚胺钒络合物的催化作用下,成功实现乙烯与该种含乙氧基硅烷极性单体的共聚,并研究了共聚产物的热性能及交联性能。
1 实验材料和方法
1.1 材料
2-吡咯甲醛,纯度98%,萨恩化学技术(上海)有限公司;2,6-二异丙基苯胺,纯度96%,萨恩化学技术(上海)有限公司;二甲基乙氧基硅烷,纯度94%,Alfa Aesar(中国)化学有限公司;正丁基锂,2.5 mol·L-1正己烷溶液,北京伊诺凯技科技有限公司;三氯化钒四氢呋喃络合物VCl3(THF)3,0.5mol·L-1二氯甲烷溶液,Acros公司;1,7-辛二烯,纯度97%,Alfa Aesar(中国)化学有限公司;Karstedt催化剂,1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷铂(0),Pt质量分数为2%的二甲苯溶液,上海阿拉丁生化科技股份有限公司,使用前可稀释至Pt质量分数为0.25%;三氯乙酸乙酯ETA,纯度97%,上海阿拉丁生化科技股份有限公司;氯化二乙基铝Et2AlCl,0.9 mol·L-1甲苯溶液,萨恩化学技术(上海)有限公司;三氟甲磺酸,99%,北京百灵威科技有限公司;乙烯,扬子石化公司,使用前需经过乙烯纯化箱纯化处理;甲醇,一级色谱纯,购自天津四友精细化学品有限公司;超干燥试剂:四氢呋喃、二氯甲烷和正己烷均购自萨恩化学技术(上海)有限公司,纯度分别为99.5%、99.9%和99.7%;其他化学试剂如甲酸、乙酸、甲苯、二甲苯、无水乙醇等均为分析纯试剂,购自国药集团化学试剂有限公司,其中用于聚合反应的甲苯溶剂需要通过溶剂纯化装置处理,除去溶解的水和氧气后方可使用。
聚合反应中所使用的催化剂[2-(2,6-iPr2C6H3N═CH)C4H3N]VCl2(THF)2吡咯亚胺钒络合物(VCIP)按照文献[19]所述的方法制备,反应路线如图1所示。
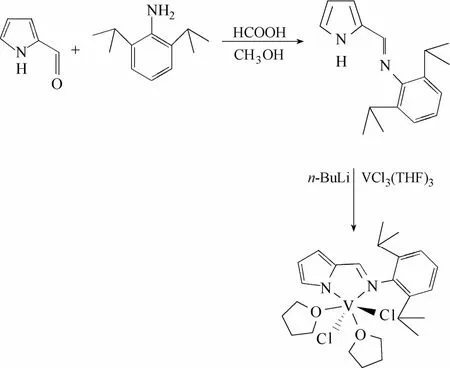
图1 吡咯亚胺钒络合物合成路线Fig. 1 Synthesis of vanadium complex bearing iminopyrrolide ligands
1.2 分析测试仪器和表征
7-辛烯基乙氧基二甲基硅烷(OEMS)的结构通过核磁共振氢谱1H NMR和碳谱13C NMR测定,温度为25℃,溶剂为氘代氯仿,采用德国Bruker Advance DMX400型核磁共振波谱仪。高聚物的组成结构由高温核磁共振氢谱1H NMR和碳谱13C NMR测定,温度为125℃,溶剂为氘代邻二氯苯,采用Bruker AC 400 脉冲型核磁共振波谱仪;高聚物的分子量和分子量分布由高温凝胶渗透色谱仪(HT-GPC)测定,型号为Viscotek 350A型,产自美国Viscotek公司,流动相为三氯苯,标样为聚苯乙烯,测试温度为150℃。聚合物热性能通过差示扫描量热仪(DSC)测定,TA Q200型,美国TA公司。温度程序为:30℃·min-1升温至150℃,保温5 min;10℃·min-1降温至30℃,保温3 min;10℃·min-1升温至150℃,选取第2、3阶段程序进行分析。
1.3 7-辛烯基乙氧基二甲基硅烷(OEMS)的制备
7-辛烯基乙氧基二甲基硅烷按照图2所示的合成路线制备。在250 ml三口烧瓶中加入16.5 g(150 mmol)1,7-辛二烯、30 g甲苯与0.215 g Pt质量分数为0.25%的Karstedt催化剂溶液,向反应体系中通入氮气保护并开启搅拌后,缓慢滴入3.1 g(30 mmol)二甲基乙氧基硅烷,滴加完毕后在50℃下继续反应5 h,注意控制1,7-辛二烯与二甲基乙氧基硅烷的反应投料摩尔比为5:1。反应完全后,通过旋蒸可除去反应体系中多余的1,7-辛二烯与甲苯溶剂;然后,过硅胶柱除去残余的少量催化剂,可得到澄清透明的油状液体即7-辛烯基乙氧基二甲基硅烷(OEMS)。
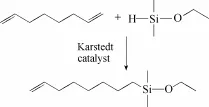
图2 7-辛烯基乙氧基二甲基硅烷合成路线Fig. 2 Synthetic route of 7-octenylethoxydimethylsilane
1.4 含功能性乙氧基硅烷聚乙烯(PE-co-OEMS)的制备
含功能性乙氧基硅烷聚乙烯(PE-co-OEMS)按照如图3所示的反应路线合成,乙烯与7-辛烯基乙氧基二甲基硅烷(OEMS)的共聚反应需在无水无氧条件下进行。反应前需为250 ml反应瓶安装机械搅拌装置并充分去除体系内水氧,然后在乙烯体系下加入适量干燥的甲苯溶剂并搅拌均匀,反应溶液的总体积为100 ml。极性单体7-辛烯基乙氧基二甲基硅烷已通过多次抽真空-氮气鼓泡法纯化。向反应体系中依次加入一定量的ETA、Et2AlCl、极性单体OEMS并搅拌均匀,反应从加入吡咯亚胺钒催化剂的甲苯溶液后开始计时,搅拌速率为500 r·min-1,反应温度为50℃,乙烯持续地通入体系并保持常压。反应10 min后,加入酸化乙醇溶液(冰醋酸与无水乙醇按照一定比例配制)终止聚合反应。产物溶液倒入酸化乙醇溶液中充分搅拌2 h,过滤、无水乙醇洗涤、干燥即可得到共聚产物。
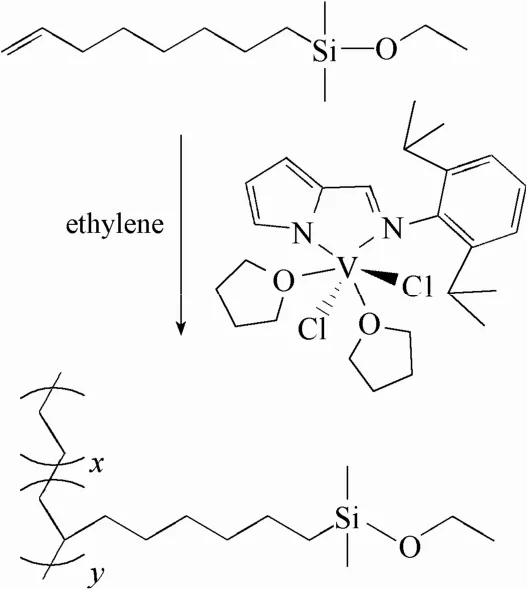
图3 乙烯与7-辛烯基乙氧基二甲基硅烷共聚反应Fig. 3 Copolymerization of ethylene and 7-octenylethoxydimethylsilane
1.5 PE-co-OEMS的交联
称取500 mg上述共聚产物PE-co-OEMS于50 ml三口烧瓶中,加入5 ml二甲苯溶液,搅拌并升温至100℃使其充分溶解。加入5 mg三氟甲磺酸,反应5 h,旋蒸除去溶剂并干燥交联产物。
1.6 交联产物凝胶含量的测定
称取交联产物(m1)于滤纸包中,在索氏提取器中用二甲苯于140℃抽提24 h,除去热溶剂,样品用丙酮洗涤并干燥至恒重(m2),交联后凝胶含量按照式(1)计算

2 实验结果与讨论
2.1 OEMS的合成及结构表征
含功能性基团的极性单体OEMS按照如图2所示的反应路线合成。在Karstedt催化剂的作用下,二甲基乙氧基硅烷与过量的1,7-辛二烯(摩尔比为1:5)反应,经过后处理,纯度高达98%。7-辛烯基乙氧基二甲基硅烷的1H NMR谱图和13C NMR谱图如图4和图5所示,图4中δ=5.72和δ=4.88处的多重峰证明碳碳双键的存在,δ=3.56处的四重峰和1.10的三重峰证明功能性乙氧基的存在;结合1H NMR谱图和13C NMR谱图可知,该单体已成功合成并可以用于后续的共聚反应。1H NMR (400 MHz, CDCl3):δ=5.72(m, 1H, ═CH—), 4.88(m, 2H,═CH2), 3.56 (q, 2H, —O—CH2—), 1.96 (m, 2H, ═CH—CH2), 1.26 (m, 8H, ═CH—CH2—(CH2)4—), 1.10 (t, 3H, —O—CH2—CH3), 0.50 (t, 2H, —Si—CH2—), 0.05 (s, 6H, —Si—CH3—)。

图4 7-辛烯基乙氧基二甲基硅烷的1H NMR谱图Fig. 41H NMR spectrum of 7-octenylethoxydimethylsilane

图5 7-辛烯基乙氧基二甲基硅烷的13C NMR谱图Fig. 513C NMR spectrum of 7-octenylethoxydimethylsilane
2.2 PE-co-OEMS的合成与结构表征
乙烯与7-辛烯基乙氧基二甲基硅烷OEMS的共聚反应按照图3的反应路线进行,反应结果如表1所示。共聚过程使用吡咯亚胺钒络合物(VCIP)作为催化剂,三氯乙酸乙酯ETA为活化剂,氯化二乙基铝Et2AlCl为助催化剂;由于该钒系催化剂对—Si—O—等极性基团具有良好的耐受性,因此极性单体OEMS在反应过程中不需要进行基团保护。
从表1可以看出,当OEMS的加料浓度为10 mmol·L-1时,共聚反应活性为5.8 kg·mmol-1·h-1,约为均聚反应活性的一半,说明极性单体的加入会对催化剂的活性中心产生一定程度的毒害作用;然而,随着极性单体浓度提高至60 mmol·L-1,该催化体系的活性仍然能够保持在1.2 kg·mmol-1·h-1,由此可见该催化剂(VCIP)可以高效催化乙烯与7-辛烯基乙氧基二甲基硅烷的共聚反应。另外,随着OEMS的加料浓度从10 mmol·L-1增加至30 mmol·L-1,PE-co-OEMS的数均分子量从8.3×103降低至7.9×103,重均分子量从35.6×103降低至16.9×103,分子量分布PDI从4.3降低至2.1。当OEMS的加料浓度超过30 mmol·L-1时,共聚产物的数均分子量、重均分子量均有所增加,且分子量分布变宽,但并未产生明显凝胶;与之前文献[31]中所报道的结果相比,本文所选取的催化体系和后处理方法能够帮助克服共聚过程中产生的凝胶。
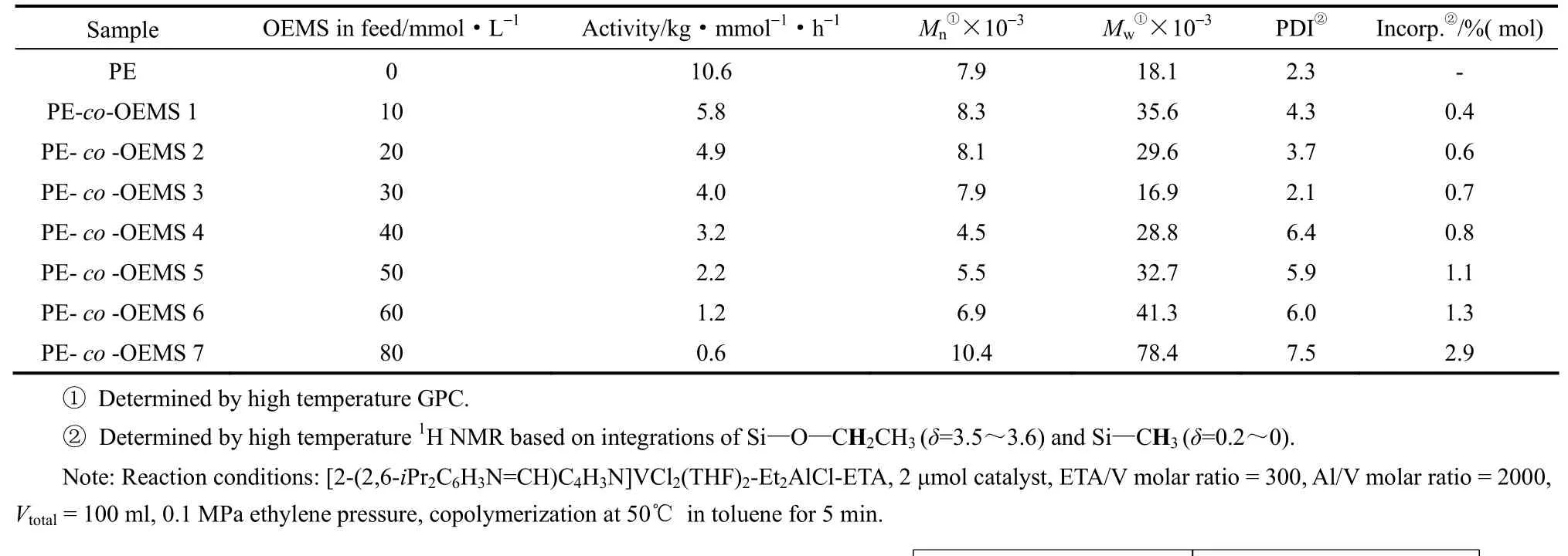
表1 乙烯与7-辛烯基乙氧基二甲基硅烷OEMS共聚反应结果Table 1 Results of copolymerization of ethylene and 7-octenylethoxydimethylsilane
PE-co-OEMS的高温1H NMR谱图和13C NMR谱图如图6和图7所示。图6中δ=0.21和δ=3.56处吸收峰证明Si—CH3和Si—O—CH2—基团的存在;图7中δ=-2.0处吸收峰也证明Si—CH3的存在,δ=58.50和δ=19.05处吸收峰证明Si—O—CH2—CH3的存在,由此可知OEMS已成功插入聚乙烯主链中。该单体的摩尔插入率可由1H NMR谱图计算得出,随着反应体系中OEMS的投料量从10 mmol·L-1增加至80 mmol·L-1,共单体的插入率从0.4%升高至2.9%(摩尔分数)。

图6 PE-co-OEMS的1H NMR谱图Fig. 61H NMR spectrum of PE-co-OEMS

图7 PE-co-OEMS的13C NMR谱图Fig. 713C NMR spectrum of PE-co-OEMS
2.3 PE-co-OEMS的热性能
表2和图8显示了PE-co-OEMS的热性能,包括结晶温度(Tc)、熔点(Tm)、结晶焓(ΔHc)、熔融焓(ΔHm)和结晶度。结果表明,随着PE-co-OEMS中硅氧烷单体单元数量的增加,结晶温度(Tc)、熔点(Tm)、结晶焓(ΔHc)、熔融焓(ΔHm)和结晶度均呈现下降趋势。当OEMS加料浓度为10 mmol·L-1时,结晶温度Tc为117.12℃,熔点Tm为130.64℃,结晶焓ΔHc为176.0 J·g-1,熔融焓ΔHm为158.2 J·g-1,结晶度为55.1%;当OEMS加料浓度提高至80 mmol·L-1时,结晶温度Tc为110.35℃,熔点Tm为120.84℃,结晶焓ΔHc为120.3 J·g-1,熔融焓ΔHm为108.1 J·g-1,结晶度为37.6%。这是由于随着共聚物中柔顺的硅烷支链的增多,对PE链段结晶的影响逐渐增大,使共聚物结晶度下降,由此导致Tc、Tm、ΔHc和ΔHm下降,如图8所示。
2.4 PE-co-OEMS的交联性能
由以上分析可知,OEMS中所含有的乙氧基在共聚反应后仍得以保留,因而可实现共聚物的后续功能化。通过共聚反应所得的PE-co-OEMS可在三氟甲磺酸的作用下快速交联,交联实验的结果如图9所示。均聚产物PE(反应体系不加入极性单体OEMS)不会发生交联,共聚产物PE-co-OEMS 3-7交联后凝胶含量≥98%;PE-co-OEMS 1-2交联后凝胶含量分别为15%和65%,这可能是由于这两组共聚产物中OEMS链段的含量较其他各组共聚产物偏低,不足以形成完全的交联产物。

表2 PE-co-OEMS的热性能Table 2 Thermal properties of PE-co-OEMS

图8 PE-co-OEMS的热性能Fig. 8 Thermal properties of PE-co-OEMS

图9 PE-co-OEMS交联后的凝胶含量Fig. 9 Gel content after crosslinking of PE-co-OEMS
3 结 论
本文报道了一种含功能性乙氧基硅烷聚乙烯(PE-co-OEMS)的制备方法,对其结构、热性能和交联性能进行了研究。
(1)通过二甲基乙氧基硅烷与1,7-辛二烯的硅氢加成反应,成功合成了功能性单体7-辛烯基乙氧基二甲基硅烷(OEMS)。
(2)在吡咯亚胺钒络合物(VCIP)的催化作用下,乙烯与OEMS配位共聚即可制得含功能性乙氧基硅烷的乙烯共聚物(PE-co-OEMS)。该共聚反应活性可保持在1.2 kg·mmol-1·h-1,且共单体OEMS的插入率可在0.4~2.9%(摩尔分数)范围内调控,共聚过程不产生凝胶。随着共单体OEMS投料量的增大,熔点(Tm)和结晶焓(ΔHc)均下降。
(3)在三氟甲磺酸作用下,OEMS单元含量较高的PE-co-OEMS交联后凝胶含量可达98%,表明该种功能化聚烯烃具有优异的可交联性。
[1] CHUM P S, SWOGGER K W. Olefin polymer technologies—history and recent progress at the dow chemical company[J]. Progress in Polymer Science, 2008, 33(8): 797-819.
[2] LOPEZ R G, D'AGOSTO F, BOISSON C. Synthesis of well-defined polymer architectures by successive catalytic olefin polymerization and living/controlled polymerization reactions[J]. Progress in Polymer Science, 2007, 32(4): 419-454.
[3] DONG J, HU Y. Design and synthesis of structurally well-defined functional polyolefinsviatransition metal-mediated olefin polymerization chemistry[J]. Coordination Chemistry Reviews, 2006, 250(1/2): 47-65.
[4] NAQVI M K, CHOUDHARY M S. Chemically modified polyolefins and their blends[J]. Journal of Macromolecular Science, Part C, 1996, 36(3): 601-629.
[5] YANJARAPPA M J, SIVARAM S. Recent developments in the synthesis of functional poly(olefin)s[J]. Progress in Polymer Science, 2002, 27(7): 1347-1398.
[6] 徐志贤, 介素云, 李伯耿. 聚烯烃与极性聚合物的嵌段与接枝[J].化学反应工程与工艺, 2015, 31(6): 522-529. XU Z X, JIE S Y, LI B G. Progress in blocking and grafting of polyolefin and polar polymer[J]. Chemical Reaction Engineering and Technology, 2015, 31(6): 522-529.
[7] 潘博. 我国聚烯烃技术的现状及趋势研究[J]. 中国化工贸易, 2013, 12(12): 25. PAN B. Study on the status and trends of polyolefin technology in China[J]. China Chemical Trade, 2013, 12(12): 25.
[8] 汪焕心. 我国高分子材料未来发展的方向[J]. 广州化工, 2012, 40(3): 1-3. WANG H X. The future direction of China's development of polymer materials[J]. Guangzhou Chemical Industry, 2012, 40(3): 1-4.
[9] 胡友良, 乔金梁, 吕立新. 聚烯烃功能化及改性:科学与技术[M].北京: 化学工业出版社, 2006: 1-10. HU Y L, QIAO J L, LÜ L X. Functionalization and Modification of Polyolefins: Science and Technology[M]. Beijing: Chemical Industry Press, 2006: 1-10.
[10] FRANSSEN N M G, REEK J N H, BAS D B. ChemInform abstract: synthesis of functional ‘polyolefins': state of the art and remaining challenges[J]. ChemInform, 2013, 42 (37): 193-210.
[11] PURGETT M D, VOGL O. Functional polymers. XLVIII. Polymerization of ω-alkenoate derivatives[J]. Journal of Polymer Science Part A: Polymer Chemistry, 1988, 26(3): 677-700.
[12] CARRICK W L. Mechanism of ethylene polymerization with vanadium catalysts[J]. Journal of the American Chemical Society, 1958, 80(23): 6455-6456.
[13] CARRICK W L, KLUIBER R W, BONNER E F,et al. Transition metal catalysts(Ⅰ): Ethylene polymerization with a soluble catalyst formed from an aluminum halide, tetraphenyltin and a vanadium halide[J]. Journal of the American Chemical Society, 1960, 82(15): 3883-3887.
[14] LEHR M H. The active oxidation state of vanadium in soluble monoolefin polymerization catalysts[J]. Macromolecules, 1968, 1 (2): 178-184.
[15] LEHR M H, CARMAN C J. Electron spin resonance evidence of inactive Ⅴ(Ⅲ) precursor to catalytically active Ⅴ(Ⅲ) in vanadium tetrachloride Ziegler catalysts[J]. Macromolecules, 1969, 2(2): 217-219.
[16] TANG L M, WU J Q, DUAN Y Q,et al. Ethylene polymerizations, and the copolymerizations of ethylene with hexene or norbornene with highly active mono(β-enaminoketonato) vanadium(III) catalysts[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2008, 46(6): 2038-2048.
[17] WU J Q, PAN L, LIU S R,et al. Ethylene polymerization and ethylene/hexene copolymerization with vanadium(Ⅲ) catalysts bearing heteroatom-containing salicylaldiminato ligands[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2009, 47(14): 3573-3582.
[18] NOMURA K, SAGARA A, IMANISHI Y. Olefin polymerization and ring-opening metathesis polymerization of norbornene by (Arylimido)(aryloxo)vanadium(Ⅴ) complexes of the type VX2(NAr) (Oar1). Remarkable effect of aluminum cocatalyst for the coordination and insertion and ring-opening metathesis polymerization [J]. Macromolecules, 2002, 35(5): 1583-1590.
[19] XU B C, HU T, WU J Q,et al. Novel vanadium(Ⅲ) complexes with bidentateN,N-chelating iminopyrrolide ligands: synthesis, characterization and catalytic behaviour of ethylene polymerization and copolymerization with 10-undecen-1-ol[J]. Dalton Transactions, 2009, 41: 8854-8863.
[20] YILG R E, YILG R I. Silicone containing copolymers: synthesis, properties and applications[J]. Progress in Polymer Science, 2014, 39(6): 1165-1195.
[21] ERBIL H Y, YAŞAR B, S ZER Ş,et al.Surface characterization of the hydroxy-terminated poly(ε-caprolactone)/poly(dimethylsiloxane) triblock copolymers by electron spectroscopy for chemical analysis and contact angle measurements[J]. Langmuir, 1997, 13(20): 5484-5493.
[22] CLARSON S J, SEMLYEN J A. Siloxane Polymers[M]. Englewood Cliffs, NJ: Prentice Hall, 1993:1-10.
[23] VASILE C. Handbook of Polyolefins[M]. New York: Marcel Dekker, Inc., 2000: 532.
[24] UOZUMI T, TIAN G, AHN C H,et al. Synthesis of functionalized alternating olefin copolymer and modification to graft copolymer by hydrosilylation[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2000, 38(10): 1844-1847.
[25] LI W, HUANG B. Synthesis of a polydimethylsiloxane macromonomer and its copolymerization with ethylene[J]. Die Makromolekulare Chemie, 1989, 190(10): 2373-2380.
[26] MUKBANIANI O, TITVINIDZE G, TATRISHVILI T,et al.Formation of polymethylsiloxanes with alkyl side groups[J]. Journal of Applied Polymer Science, 2007, 104(2): 1176-1183.
[27] GALL B T, THOMANN R, M LHAUPT R. Synthesis and characterization of semicrystalline triblockcopolymers of isotactic polystyrene and polydimethylsiloxane[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2011, 49(11): 2339-2345.
[28] 金震, 范宏. 长支链型PDMS-g-PE共聚物的制备及其增塑润滑作用[J]. 化工学报, 2016, 67(2): 672-678. JIN Z, FAN H. Synthesis, characterization and lubricating effect of long chain branched polydimethylsiloxane-g-polyethylene (PDMS-g-PE) copolymers[J]. CIESC Journal, 2016, 67(2): 672-678.
[29] CIOLINO A E, BARRERA GALLAND G, VILLAR M A. Novel synthesis of polyethylene-poly(dimethylsiloxane) copolymers with a metallocene catalyst[J]. Journal of Polymer Science Part A: Polymer Chemistry, 2004, 42(10): 2462-2473.
[30] JIN Z, FAN H, LI B G,et al. Synthesis of a novel type of octyltetramethyldisiloxane-containing olefinic macromonomer and its copolymerization with ethylene[J]. Polymer, 2016, 83: 20-26.
[31] ZIMMER S, SCH BEL A, HALBACH T,et al. New curable propylene copolymers containingtert-butoxysilane side groups[J]. Macromolecular Rapid Communications, 2013, 34(3): 221-226.
Preparation of functional ethoxysilane-containing polyethylene through ethylene coordination copolymerization catalyzed by iminopyrrolide vanadium complex
LIU Zhiying, FAN Hong
(State Key Laboratory of Chemical Engineering,College of Chemical and Biological Engineering,Zhejiang University,Hangzhou310027,Zhejiang,China)
A well-defined silane-functionalized comonomer, 7-octenylethoxydimethylsilane (OEMS), was synthesized through hydrosilylation reaction of dimethylethoxysilane with 1,7-octadiene. Catalyzed by a vanadium complex bearing iminopyrrolide ligands (VCIP), the functional ethoxysilane-containing polyethylenes (PE-co-OEMS) were synthesized by copolymerization of ethylene with OEMS. The structure and properties of PE-co-OEMSs were characterized with1H NMR,13C NMR, HT-GPC and DSC. It was found that catalytic activity still remained 1.2 kg·mmol-1·h-1when OEMS feeding reached to 60 mmol·L-1, and the OEMS incorporating in the copolymer was 1.3%(mol). Besides, the OEMS feeding had a great influence on the average molecular weight (Mw) and distribution (PDI) of copolymers. TheMwand PDI of PE-co-OEMS were 16900 and 2.1 when OEMS feeding was 30 mmol·L-1. With further increasing the OEMS feeding,Mwincreased and PDI became wider. The melting temperature (Tm) and crystallization enthalpy (ΔHc) decreased with the increase of OEMS incorporation. The ethoxysilane-containing copolymers were easily crosslinked under the catalysis of trifluoromethanesulfonicacid, and the copolymer gel contents after crosslinking were nearly 100%.
polyethylene; silicone; crosslinking; polymer; preparation; synthesis
Prof. FAN Hong, hfan@zju.edu.cn
TQ 325.1
:A
:0438—1157(2017)02—0774—07
10.11949/j.issn.0438-1157.20160906
2016-07-04收到初稿,2016-10-25收到修改稿。
联系人:范宏。
:刘智颖(1992—),女,硕士研究生。
国家自然科学基金重大国际(地区)合作研究项目(21420102008)。
Received date: 2016-07-04.
Foundation item: supported by the Major International (regional) Joint Research Project of the National Natural Science Foundation of China (21420102008).
