MgH2储氢体系加氢反应机理的第一性原理研究
2017-02-27张国英麻洪吉
张国英, 麻洪吉
(沈阳师范大学 物理科学与技术学院, 沈阳 110034)
MgH2储氢体系加氢反应机理的第一性原理研究
张国英, 麻洪吉
(沈阳师范大学 物理科学与技术学院, 沈阳 110034)
采用基于密度泛函理论的第一性原理方法研究了稀土和过渡元素替代对MgH2储氢体系的加氢反应影响机理。研究发现氢原子可以在Mg表面形成稳定的吸附。稀土和过渡元素Fe,V,Ti,Nb,La替代Mg降低了H的吸附能,表明合金元素可以提高H在Mg表面吸附的稳定性。表面吸附体系稳定性由高到低的次序为:Nb,Fe,V,Ti,La掺杂Mg表面。电子结构及电荷布局分析结果表明,稀土La与H间电子耦合较弱;而过渡金属原子明显与H有较强的共价相互作用。H能稳定的吸附在Mg的表面,为H进一步向Mg体内扩散提供了条件,从而为Mg氢化生成MgH2提供了H源。此外,吸附H也可能会与掺杂元素生成氢化物(如TiH2),这些氢化物也是Mg氢化生成MgH2的催化剂。合金原子与H间存在的强的共价作用是提高MgH2的加氢性能的决定原因。
MgH2储氢材料; 第一性原理; 表面吸附; 加氢反应机理
氢是一种理想的绿色清洁能源,被誉为21世纪的绿色能源载体[1],氢的储存方式有气态储氢、液态储氢、固态储氢[2],因固态储氢较气态和液态具有储氢量大、安全性高、运输方便等优点而引起了国内外学者的广泛研究。近年来,人们把工作重点放在了镁基储氢材料的研究上,我国具有丰富的镁资源,也是原镁出产大国和出口大国[3],金属镁作为储氢材料具有很多优点[4],密度小、储氢量高、资源丰富、价格低廉。然而,MgH2吸/放氢速率慢、温度高、形成的氢化物稳定、材料粉化严重,吸/放氢循环性差、易氧化等问题限制了它的实际应用。发展一种储氢材料,其可逆性是一关键因素。为改善MgH2的可逆储放氢性能,近年来人们对MgH2的吸放氢过程进行了广泛的研究。
实验发现,过渡金属、过渡金属氟化物及氧化物对MgH2储氢材料的储放氢是良好的催化剂[5-14]。Jin等[15]研究了过渡金属氟化物对储氢材料MgH2吸放氢性能的影响,发现许多氟化物对MgH2的放氢温度和吸氢动力学性能都有催化作用,其中,NbF5,TiF5可明显改善MgH2的加氢动力学性能。Rohit等[16]发现MgH2与5%的Ti,Fe,Ni进行机械球磨,产生MgH2-Ti5Fe5Ni5复合物,球磨后的解离温度与纯的MgH2相比降低了90 ℃。270 ℃时,MgH2-Ti5Fe5Ni5复合物在15 min内吸氢量5.3%,而相同条件下纯的MgH2吸氢量为4.2%。Rohit等[17]利用机械球磨技术生成MgH2-ZrFe2Hx纳米复合材料,发现包含ZrFe2和ZrFe2Hx的纳米复合材料导致解离温度从410 ℃分别降低到350 ℃和290 ℃,复合材料的加氢活化能与纯的MgH2相比降低了30 kJ/mol。Wang等[18]利用球磨方法产生的复合材料Mg-TiO2拥有高吸氢能力、动力学性能好、反应温度低且有良好的抗氧化性能。Liang等[19]通过机械合金化制备的MgH2-V复合材料,吸放氢性能较好。在储氢材料中,稀土卤化物也是被广泛添加的催化剂。Ismail等[20]研究了LaCl3对MgH2吸放氢性能的影响,发现添加10% LaCl3,MgH2初始放氢温度大约在300 ℃,比纯MgH2降低50 ℃;掺杂LaCl3的MgH2样品在320 ℃,5 min内放氢4.2%, 而纯MgH2样品在相同条件下只放氢0.2%。在加氢过程中,掺杂LaCl3的MgH2样品在300 ℃,2 min内吸氢5.1%, 而纯MgH2样品在相同条件下只吸氢3.3%。吴广新等[21]采用第一性原理方法系统研究H2分子和H原子在Mg (0001)面的吸附行为,为H2分子的解离和氢化物的放氢过程均为速控步骤这一结论提供了理论支持。综上所述,对MgH2吸氢过程大多数的研究都集中于实验探究,对加氢机理的理论研究还不够深入。因此,本文意在运用基于密度泛函理论的第一性原理方法在理论上探究过渡金属及稀土元素对储氢材料MgH2在吸氢过程中的催化影响机理,为合理设计和改善MgH2储氢材料性能提供理论指导。
1 计算模型和理论方法
1.1 晶体结构
Mg为六角密集结构,晶格常数为a=0.321 nm,c/a=1.624[22]。采用周期性边界的超晶胞模型进行表面模拟。建立了Mg的(0001)表面模型,表面超晶胞包括6层原子,表面的真空层都设为10 Å,表面结构取p(2×2)晶胞结构。为了研究MgH2储氢材料的加氢影响机理,将H吸附在Mg(0001)表面最稳定的fcc穴位上[21],用Nb,Fe,Ti,V,La替代Mg(0001)表面中的同一个Mg原子,图1(a)为6层Mg(0001)表面原子结构模型,图1(b)为H吸附在fcc穴位模型,图1(c)为不同元素(如La)替换表面镁原子后吸附H的Mg(0001)表面模型。
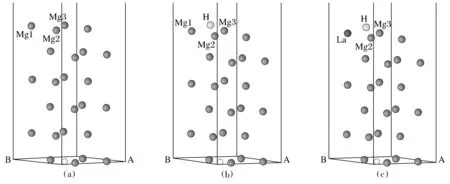
图1 本文采用的计算结构模型
1.2 计算方法
采用基于密度泛函理论的赝势平面波方法(Castep软件包)[23], 电子交换相互作用采用广义梯度近似GGA下的PBE[24]泛函描述。采用对正则条件进行弛豫的超软赝势[25]作为平面波基集。自洽叠代(SCF) 方法进行计算时, 采用结合BFGS 共轭梯度方法的Pulay 密度混合方案[26]处理电子弛豫。先对模型的晶体结构进行完全的几何优化,以求得它们的局域最稳定结构。优化结束时,体系总能量的收敛值取2.0×10-5eV/atom,作用每个原子上的力低于0.05 eV/nm,公差偏移小于2.0×10- 4nm,应力偏差小于0.1 GPa。
2 计算结果与讨论
2.1 H吸附-MgH2生成
首先来研究MgH2的生成机理。H稳定吸附是MgH2生成的先决条件。因此计算纯净和不同元素替换前后Mg(0001)表面系统吸附能,以此讨论含这些元素的催化剂对MgH2的加氢反应的影响机理。吸附能的计算公式为:E吸附能=[EH/Mg(0001)-(EMg(0001)+EH)],其中EH/Mg(0001)是一个氢原子吸附于Mg(0001)面的总能,EMg(0001)为纯净6层Mg(0001)表面超晶胞的总能。EH由Etot(H2)/2 给出。Etot(H2)是通过优化一个8个顶角各有一个H2的边长为1 nm的立方体来确定的,H2的键长取0.074 9 nm。负值的吸附值表示吸附体系比单独的组分体系稳定,且负值越小,吸附氢原子的体系就越稳定。
表1为Mg表面存在替代元素后H的吸附能,发现吸附能均为负值,表明氢原子可以在Mg表面形成稳定的吸附。比较各替代元素的影响,发现Fe,V,Ti,Nb,La替代后,H的吸附能均比未替代前小,表明元素替代后H与Mg表面形成了更为稳定的吸附体系,掺杂元素表现出了更强的亲氢性。Nb替换后的吸附体系最为稳定。表面吸附体系稳定性由高到低依次为Nb,Fe,V,Ti,La。与纯净的Mg表面相比,La掺杂也提高了H吸附的稳定性,但较其他的元素的影响要小的多。总之,过渡金属和稀土元素的添加提高了H吸附的稳定性,即改善了MgH2的加氢性能。

表1 H在纯Mg和掺杂Mg表面的吸附能
2.2 电子结构分析
为了更好地理解H在Mg表面吸附生成MgH2及掺杂元素的影响的机理,计算了H原子吸附于纯净及元素掺杂Mg(0001)表面各原子的态密度和电子差分密度,见图2、图3。态密度和电子差分密度对于分析材料中的原子成键和材料特性有重要的意义。图2分别为纯净Mg表面(a)Mg1、Mg2原子、掺杂Mg表面((b)掺杂La;(c)掺杂V;(d)掺杂Ti;(e)掺杂Fe;(f)掺杂Nb)Mg2及掺杂原子各表面吸附的H原子的分波态密度及吸附体系的总态密度。
由图2(a)可以看出,Mg表面原子s,p轨道电子都对总态密度有贡献。Mg的p电子与H的s电子有一定的作用;在-6 eV附近存在Mg与H原子的s-s电子共价作用的峰,但这个峰值很小(约0.3),表明Mg与H的共价作用较弱。观察元素替代后(图2(b)~图2(f))各原子的分波态密度图,可以看出,Nb,Ti,Fe,V,La的替换均在费米能级附近引入一态密度峰,这一峰来自于它们d电子贡献。观察到Mg在过渡元素或稀土d电子引起的峰值位置,Mg的p电子态也存在一个峰值,说明Mg的p电子有共价作用。再看过渡元素或稀土与H的作用,在-6 eV附近存在过渡元素或稀土与H原子的s-s电子共价作用的峰,峰值也不高。此外过渡元素或稀土的d电子也与H的s电子有一定的共价作用。
为了更形象更直观的描绘出H与表面Mg及掺杂原子的共价作用,图3给出了电子差分密度图。
从图3可以看出,体内的Mg原子,电子云是球形分布的,主要是s电子的贡献。而吸附H的Mg表面,Mg原子的电子密度等值线已不是球形,是哑铃形,说明有p电子贡献。再看过渡元素或稀土元素替代后的H吸附表面,过渡元素或稀土元素的电子密度图明显是d电子贡献的十字花瓣形。再仔细观察掺杂后吸附H原子电子密度的变化:La掺杂时,H电子密度形状变为椭球形,说明H与La有一定的电子耦合;V,Ti,Nb,Fe掺杂时,掺杂原子明显与H有电子云交叠,说明具有较强的共价相互作用。这使得H能稳定的吸附在Mg的表面,为H进一步向Mg体内扩散提供了条件,从而为Mg氢化生成MgH2提供了H源。此外,吸附H也可能会与掺杂元素生成氢化物如TiH2,这些氢化物也是Mg氢化生成MgH2的催化剂。
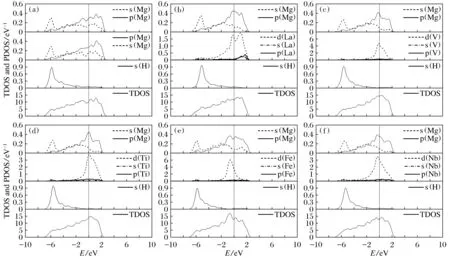
图2 总态密度和各原子的分波态密度
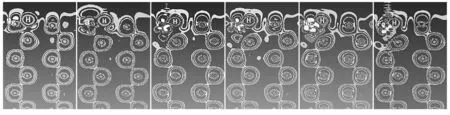
图3 替换前后电子的差分密度图(从左至右依次Mg,La,V,Ti,Nb,Fe)
2.3 电荷布局及重叠布局
电子差分密度图形象地给出了Mg表面体系所成共价键的性质,但还无法进行量化分析;此外原子间的离子键结合性质无法反映出来。为此对纯净及掺杂Mg表面各原子的电荷得失,各键的重叠布局及键长进行了计算,见表2和表3。
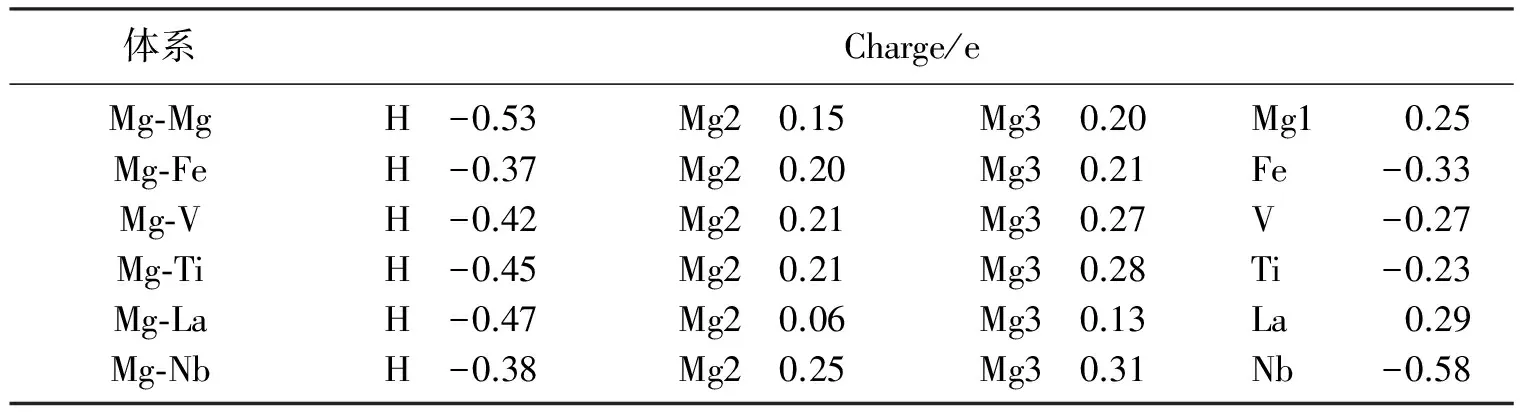
表2 元素替换前后Mg表面各原子电子得失
从表2(表的第1行)可以看出,在纯Mg表面,Mg失电子,H得电子,可见Mg与H之间存在较强的离子键。Nb,V,Ti,Fe,La合金元素替代Mg1后,H得到的电子数减少。除稀土La失电子外,其他合金元素由于存在空的d轨道也得电子,因此H与La静电吸引,而其他合金元素与H存在静电排斥,Mg与H之间存在静电吸引。从图3可知,合金元素替代Mg1以后,H原子都移向合金原子,而合金原子除La外都静电排斥H,可见静电作用在原子间相互作用中不占主导作用。
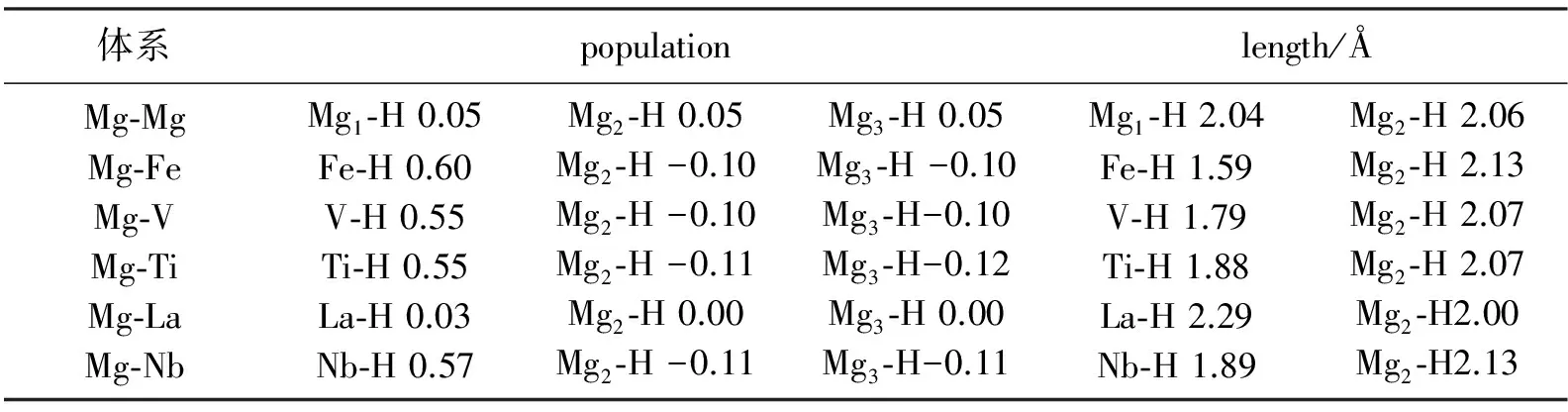
表3 体系中原子间的重叠布局数及键长
从表3的第1行可以看到,Mg与H之间重叠布局数很小,说明其间的共价作用较小。所以在Mg表面吸附H与Mg之间主要是靠离子键结合的。La合金元素替代Mg1后,La与H间的重叠布局数很小,Mg与H间的也很小,可见稀土元素对氢吸附的影响较小,因此其对MgH2的加氢行为影响也不大。Nb,V,Ti,Fe元素取代Mg1后,合金原子与H间的重叠布局明显增大,可见合金原子与H间的共价作用很强,这可能是合金元素提高H吸附稳定性,生成氢化物催化剂的原因。再看合金元素对键长的影响,La与H间的键长比纯Mg表面Mg-H间的键长还长,可见La对氢吸附影响不大;Nb,V,Ti,Fe元素取代Mg1后,合金原子与H间的键长明显变短,这与图3的结果一致。这也说明合金原子与H间共价作用强。合金原子与H间存在较强的共价作用可能是提高MgH2的加氢性能的决定原因。
3 结 论
应用基于密度泛函理论的第一性原理方法,研究了MgH2储氢体系的加氢反应机理。分析可得:氢原子可以在Mg表面形成稳定的吸附。Fe,V,Ti,Nb, La替代Mg降低了H吸附能,表明替代元素可以提高H在Mg表面吸附的稳定性。表面吸附体系稳定性由高到低依次为:Nb,Fe,V,Ti,La掺杂Mg表面。电子分析表明,稀土La与H间电子耦合较弱;而过渡金属原子明显与H有较强的共价相互作用。H能稳定的吸附在Mg的表面,为H进一步向Mg体内扩散提供了条件,从而为Mg氢化生成MgH2提供了H源。此外,吸附H也可能会与掺杂元素生成氢化物如TiH2,这些氢化物也是Mg氢化生成MgH2的催化剂。合金原子与H间存在的强的共价作用是提高MgH2的加氢性能的决定原因。
[ 1 ]SCHLAPBACH L, ZÜTTEL A. Hydrogen-storage materials for mobile applications[J]. Nature, 2001,414:353-358.
[ 2 ]陈加福,陈志民,许群. 绿色能源----氢气及无机材料储氢的研究进展[J]. 世界科技研究与发展, 2007,29(5):32-38,56.
[ 3 ]师昌绪,李恒德,王淀佐,等. 加速我国金属镁工业发展的建议[J]. 材料导报, 2001,15(4):5-6.
[ 4 ]汪云华,王靖坤,赵家春,等. 固体储氢材料的研究进展[J]. 材料导报, 2011,25(9):120-124.
[ 5 ]张健,孙立芹,毛聪,等. Ni、Ti单独/复合掺杂对MgH2组织结构与解氢性能的影响及机理[J]. 材料导报, 2015,29 (22):91-95.
[ 6 ]腊明,陈昌东,程昊. Sc对镁基储氢材料吸放氢动力学影响[J]. 稀土, 2016,37(3):90-95.
[ 7 ]张健,朱璞,毛聪,等. 碳材料掺杂对镁基氢化物释氢性能的影响及其微观机理[J]. 中国有色金属学报, 2015,25(9):2464-2470.
[ 8 ]朱玥,李永成,王福合. Li掺杂对MgH2(001)表面H2分子扩散释放影响的第一性原理研究[J]. 物理学报, 2016, 65(5):254-259.
[ 9 ]袁江,周惦武,魏红伟. TiF3对MgH2体系解氢热力学影响的第一性原理研究[J]. 中国有色金属学报, 2016,26(7):1480-1486.
[10]YU H,BENNICI S,AUROUX A. Hydrogen storage and release: Kinetic and thermodynamic studies of MgH2activated by transition metal nanoparticles[J]. International Journal of Hydrogen Energy, 2014,39(22):11633-11641.
[11]HUSSAIN T,MAARK T A,PATHAK B, et al. Improvement in the hydrogen desorption from MgH2upon transition metals doping: A hybrid density functional calculations[J]. AIP Advances, 2013,3(10):102-117.
[12]孙海全,邹建新,曾小勤,等. 纯Mg及Mg-Nd超细粉体的制备及其储氢性能[J]. 稀有金属材料与工程, 2012,41(10):1819-1823.
[13]ZHOU C S,FANG Z Z,REN C,et al. Effect of Ti intermetallic catalysts on hydrogen storage properties of magnesium hydride[J].J Phys Chem C, 2013,117(25):12973-12980.
[14]张健,黄雅妮,毛聪,等. Ti,V,Nb掺杂MgH2储氢体系的放氢性能及微观机理[J]. 化学学报, 2010,68(20):2077-2085.
[15]SEON-AH J, JAE-HYEOK S, CHO Y W, et al. Dehydrogenation and hydrogenation characteristics of MgH2with transition metal fluorides[J]. Journal of Power Sources, 2007,172(2):859-862.
[16]SHAHI R R, TIWARI A P, SHAZ M A, et al. Studies on de/rehydrogenation characteristics of nanocrystalline MgH2co-catalyzed with Ti, Fe and Ni[J]. International Journal of Hydrogen Energy, 2013,38(6):2778-2784.
[17]SHAHI R R, BHATANAGAR A, PANDEY S K, et al. MgH2-ZrFe2Hxnanocomposites for improved hydrogen storage characteristics of MgH2[J]. International Journal of Hydrogen Energy, 2015,40(35):11506-11513.
[18]WANG P, WANG A M, ZHANG H F, et al. Hydrogenation characteristics of Mg-TiO2(rutile) composit[J]. Journal of Alloys and Compounds, 2000,313(1/2):218-223.
[19]LIANG G, HUOT J, BOILY S, et al. Hydrogen desorption kinetics of a mechanically milled MgH2+5%V nanocomposite[J]. Journal of Alloys and Compounds, 2000,305(1/2):239-245.
[20]ISMAIL M. Effect of LaCl3addition on the hydrogen storage properties of MgH2[J]. Energy, 2015,79(1):177-182.
[21]吴广新,张捷宇,吴永全,等. H在Mg(0001)表面吸附、解离和扩散的第一性原理研究[J]. 物理化学学报, 2008,24(1):55-60.
[22]KITTEL C. Introduction to solid state physics[M]. New York:Willey, 1986:194.
[23]LINDAN P J D. First principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys Condens Matter, 2002,14(11):2717-2744.
[24]PERDEW, BURKE, ERNZERHOF. Generalized gradient approximation made simple[J]. Physical review letters, 1996,77(18):3865-3868.
[25]VANDERBILT D. Soft self consistent pseudopotentials in a generalized eigenvalue formalism[J]. Phys Rev B, 1990,41(11):7892-7895.
[26]HAMMER B, HANSEN L B, NORKOV J K. Improved adsorption energetics within density functional theory using revised Perdew Burke Ernzerh of functionals[J]. Phys Rev B, 1999,59(11):7413-7421.
First-principles study on the hydrogenation mechanisms of MgH2hydrogen storage materials
ZHANGGuoying,MAHongji
(College of Physics Science and Technology, Shenyang Normal University, Shenyang 110034, China)
The pseudopotential plane-wave method based on density functional theory was used to study the influences mechanisms of the transition metal elements and rare earth substitution on the hydrogenation of MgH2. The results show that H atom can be adsorbed on the Mg(0001) surface stably. The substitution of rare earth and transition elements Fe, V, Ti, Nb, La for Mg in the Mg surface decreases the adsorption energies of H, which shows that the alloying element can improve the stability of H adsorption on Mg surface.The ranking of the stability of the surface adsorption system from high to low is: Nb,Fe,V,Ti,La doped Mg surface. The analysis results of electronic structure and charge population show that the electron coupling between La and H is weak; while the covalent interaction between transition metal atom and H is obviously stronger. The stable adsorption of H on the surface of Mg provides the condition for H to further diffuse into the bulk Mg, and provides a H source for Mg hydrogenation to produce MgH2. In addition, adsorption of H may also be related to the formation of hydrides (such as TiH2). Such hydrides are also the catalysts in Mg hydrogenation to produce MgH2. A strong covalent interaction between alloy atoms and H is a crucial factor to improve hydrogenation performance of MgH2.
MgH2hydrogen storage system; first-principle; surface adsorption; the mechanism of hydrogenation reaction
2016-09-12。
国家自然科学基金资助项目(51371049); 国家高技术研究发展计划(“863”计划)( 2009AA05Z105)。
张国英(1965-),女,辽宁沈阳人,沈阳师范大学教授,博士。
1673-5862(2017)01-0023-06
TG139.7
A
10.3969/ j.issn.1673-5862.2017.01.004
