Ni离子掺杂锐钛矿相TiO2体系的第一性原理研究
2017-02-16于智清王逊田昂刘艳侠杨合薛向欣
于智清王逊田昂刘艳侠杨合薛向欣*,
(1东北大学冶金学院,沈阳110819)
(2辽宁省冶金资源循环科学重点实验室,沈阳110189)
(3沈阳建筑大学理学院,沈阳110168)
(4辽宁大学物理学院,沈阳110036)
(5中国科学院金属研究所钛合金研究部,沈阳110016)
Ni离子掺杂锐钛矿相TiO2体系的第一性原理研究
于智清1,3王逊3田昂1,2刘艳侠4,5杨合1,2薛向欣*,1,2
(1东北大学冶金学院,沈阳110819)
(2辽宁省冶金资源循环科学重点实验室,沈阳110189)
(3沈阳建筑大学理学院,沈阳110168)
(4辽宁大学物理学院,沈阳110036)
(5中国科学院金属研究所钛合金研究部,沈阳110016)
采用自旋密度泛函理论的第一性原理方法并结合晶体配位场理论,研究了Ni离子掺杂锐钛矿相TiO2体系(NixTi1-xO2;NixTiO2)的几何结构、缺陷形成能、电子结构以及磁性特征等问题。结果表明:实验上发现的有关Ni掺杂TiO2体系的很多矛盾,如:晶粒体积的增减、掺杂Ni离子的不同价态、吸收光谱带边移动方向和体系磁性特征的差异等问题都可归因于Ni离子掺杂类型的不同(NiTi;Niin)。分析表明,不同的Ni离子掺杂类型导致所成Ni-O键的键长和电荷布居不同,从而使Ni离子呈现不同的价态,这也是体系宏观电学和磁学性能差异的根本原因。形成能计算表明,通过控制Ni-TiO2晶体生长过程中的氧环境,可以人为的控制Ni离子的掺杂类型。
镍掺杂;第一性原理;电子结构;形成能
0 引言
TiO2作为一种性能优良的光催化材料,由于具有高经济性、低毒性、良好的生物相容性和耐久性等特点而被广泛关注[1]。近年来,随着对其研究的深入,TiO2材料的应用范围也不断拓展。比如:光防腐领域的研究发现,覆盖在铜、不锈钢等金属表面的TiO2薄膜在发生光电跃迁时,进入TiO2导带的电子会迁移进入金属表面,从而帮助金属抵御海水等物质的腐蚀[2-4];气体探测领域的研究发现,O2、NH3、H2、SO2等气体分子很容易吸附在具有较高比表面积的TiO2纳米管阵列(TNTA)之上,并改变TNTA的导电特性[5]。所以TiO2气体传感器对此类气体具有较高的探测效率。此外,在新一代阻变式存储器(ReRAM)材料的研制过程中,TiO2也被寄予厚望[6]。但是,纯TiO2材料在实际应用过程中也有很多问题需要解决。例如:作为光催化和光防腐材料,TiO2材料的可见光利用率较低而光生电子-空穴(e-/h+)的复合率较高[7-8];作为ReRAM材料,TiO2内部会随机形成不规则的导体丝,从而改变元件的切换参数[9]。
研究表明,利用过渡金属离子对TiO2材料进行掺杂改性可以有效提高材料的使用性能。Choi等报道[10],利用Fe3+、Mo5+、Ru3+、Os3+、Re5+、V4+、Rh3+、Rh3+等离子的掺杂都能在TiO2本征禁带之中引入杂质能级从而有利于光生电子的带间跃迁,并抑制e-/h+的复合。TiO2基ReRAM材料的研究也发现,3d过渡金属Cu、Ni、Co、Cr、V等掺杂可以控制导电丝的形成,从而调节元件内部的导电特性[9]。理论方面,Zhao等利用投影缀加波法(projected-augmented wave method,PAW)的研究表明V、Cr、Co、Ni、Cu等元素的掺杂都会在禁带之中引入杂质能级,且掺杂离子价电子数越多,杂质能级出现的位置越深[9]。柳宗彦等对3d过渡金属离子掺杂TiO2的光吸收系数进行了对比,结果表明Cr、Mn、Fe、Co、Ni、Cu等元素的掺杂都可以使的TiO2光吸收带边红移,从而提高可见光利用率[11]。
在众多的过渡金属离子之中,利用3d过渡金属Ni对本征TiO2进行掺杂改性,是近年来研究的热点之一。这主要是由于,Ni2+和Ti4+的离子半径相似[4],Ni2+较容易进入TiO2晶格之中而形成稳定的掺杂体系。比如:Yao等利用等离子体电解氧化法,制备了厚度为1μm的Ni掺杂TiO2薄膜,并用其催化分解Na2S+Na2SO3溶液,发现Ni掺杂可以使吸收光谱带边明显红移,且Ni掺杂量为2.45%(物质的量分数)时的催化效率最高[12]。Visinescu等也用直流磁控溅射法制备了Ni掺杂TiO2薄膜,发现Ni掺杂使薄膜的丙酮降解能力提升了60%[13]。Pol等发现,Ni掺杂的TiO2颗粒对人工染料罗丹明B的还原能力大幅提高,并且Ni掺杂使体系在室温下具有磁性[14]。Sun等报道,在304不锈钢表面覆盖Ni掺杂量为0.5%(物质的量分数)的TiO2涂层,将使金属的防腐性能得到较大的提升[4]。
虽然理论上对Ni掺杂TiO2也进行了许多研究[11,15-16],但还不够深入。比如,实验证明,Ni有两种方式进入TiO2晶格之中[4]:(i)Ni替代晶格Ti形成的替位掺杂NiTi;(ii)Ni离子进入TiO2晶格间隙之中形成的晶隙掺杂Niin。但理论上关于Ni晶隙掺杂的计算还未见报道。实验上关于掺杂Ni离子的价态问题也有很多矛盾,多数报道认为掺杂Ni为稳定性最高的Ni2+,但这无法解释掺杂体系的磁性问题[17-19]。此外,掺杂导致的晶胞体积变化等问题也有很多争议[12]。这都需要更加深入的理论计算和分析。为了解决上述问题,本文运用第一性原理方法,系统的分析了掺杂量、掺杂方式、晶体场构型等对Ni掺杂TiO2材料的影响,这将对实验研究提供有益的参考。
1 计算方法
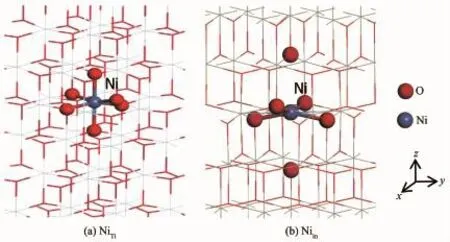
图1 NiTi和Niin掺杂体系Fig.1Structure of the Ni doping anatase
本文计算采用基于自旋密度泛函理论(DFT)框架下的平面波超软赝势法。用广义梯度近似加排斥能修正(generalized gradient approximation+Hubbard U,GGA+U)的PBE方法处理电子间交换关联能。采取+U计算是对传统DFT计算结果的修正,不同文献对U值的确定方法也不尽相同,合理的Hubbard U值不仅能改进体系禁带宽度的计算,更重要的是能够得到合理的体系电子结构[20],本文计算采用近期文献的建议值(UTi=4eV,UNi=3.5eV)[21-22],所采用文献计算方法与本文相似,且都取得了良好的计算效果。由于实验发现锐钛矿相TiO2具有更好的光催化和光防腐性能[23],本文也以此为研究对象。锐钛矿相TiO2属于四方晶体结构,空间群为I41/amd,每个晶胞包含12个原子。为计算不同Ni掺杂浓度对晶体结构、电子结构等性能的影响,分别构建了2×2× 1和2×2×2超晶胞。Ni物质的量分数掺杂量为Niin:1.03%、2.04%和NiTi:1.04%、2.08%,符合实验的实现范围,并可避免Ni团簇等因素的影响[14,17]。如图1(a)、(b),用1个Ni替换TiO2超晶胞中的1个晶格Ti来模拟替位掺杂NiTi;模拟晶隙掺杂Niin时,为减小晶格畸变并获得最稳定的掺杂结构[7,24],把1个Ni原子置于TiO2超晶胞z方向中心对称轴上的晶格空隙位置(分数坐标:2×2×1超晶胞(0.5x,0.5y,0.5z);2×2×2超晶胞(0.5x,0.5y,0.25z))。为保证理论计算精度和不同体系的能量可比性,所有体系的倒空间抽样间隔都设为0.3nm-1,k网格分别为4×4×4(2×2×1超胞)、4×4×4(2×2×2超胞)[24]。平面波截断能量设为400eV。自洽场收敛标准设置为5×10-7eV/atom。全部计算在CASTEP程序包中进行。
2 结果与讨论
2.1 几何结构分析
锐钛矿相TiO2中每个Ti离子被最近邻的6个O离子包围,形成TiO6八面体结构,晶体中有2种Ti-O键长deq和dap。计算所得晶格参数与实验值[25]最大偏差小于3%,说明了计算方法的合理性。计算所得键长、折合每TiO2体积、键布居值等结果列于表1。
分析表1数据发现,Ni替代晶格Ti离子形成替位掺杂NiTi时:Ni与6个最近邻的O形成Ni-O键;Ni-O键长小于被取代的Ti-O键,说明掺杂发生时,NiO6八面体中的配位O原子向中心Ni靠近,这会导致Ni3d轨道受到晶体场排斥作用更强。所以此时Ni3d轨道分裂能会大于Ti。此时的Ni-O键平均布居值小于被取代的Ti-O键(Ni-O:0.27,Ti-O:0.57),说明成键共价性减弱,这会导致晶体稳定性降低。需要注意的是,Ni2+离子的半径(0.069nm)大于被取代的Ti4+离子半径(0.0605nm[4]),而Ni-O键长度却小于被取代的Ti-O键,而且Ni-O键共价性更低;另外,掺杂形成的Ni-O键长度也比NiO晶体中的Ni-O(0.2094nm)键小很多。这说明掺杂Ni离子应以高价态Nix+(x>2)形式存在,具体的价态情况将在下面的电子结构计算中分析。数据也表明,替位掺杂浓度越高,折合晶胞体积越小。此时晶粒体积减小、比表面积增加、量子效应增强。这有利于提高粒子的活性[7]。
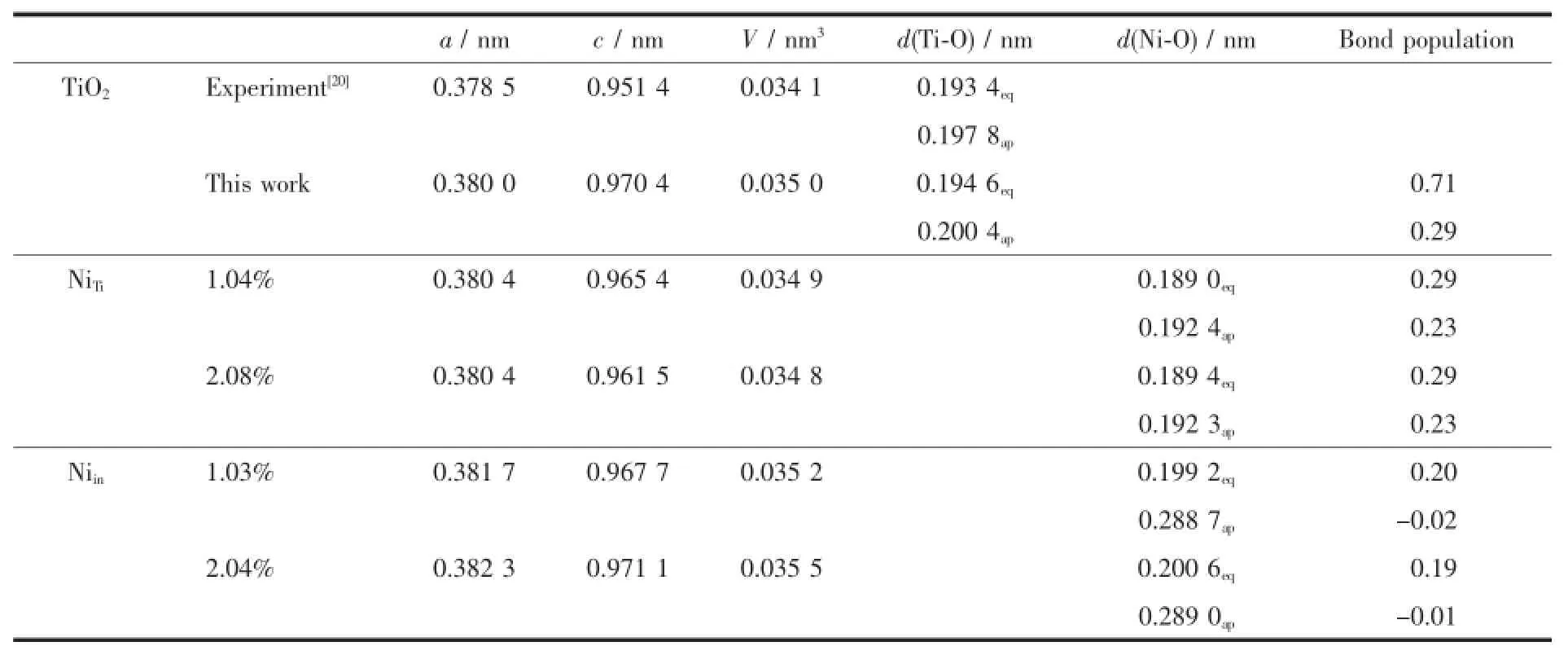
表1 几何优化后的键长、折合TiO2体积和键布居值Table 1Optimized structural parameters and bond population values of doped system
当Ni进入TiO2的晶格间隙形成晶隙掺杂Niin时:Ni处于被拉长的NiO6八面体中心,Ni与八面体xy平面的4个O原子距离较近,但与z方向的O原子距离过远。配位O原子受周围的3个成键Ti原子的束缚,没有明显的向掺杂Ni离子靠近。此时的Ni-O键长度都大于Ti-O键,Ni-O布居值也明显小于的Ti-O布居值,说明成键强度和共价性都较弱。尤其是在z方向,Ni-Oap键长(0.2887nm,0.2890nm)已经远大于正常的Ti-Oap键长(0.2004nm),Ni-Oap键布居值也变为绝对值很小的负值,说明形成微弱的反键。由于Niin的过程中会向体系引入过剩的电子,晶体库伦排斥作用增强,晶胞体积增加,这也会降低晶体的稳定性。需要说明的是,晶隙掺杂时的NiO6八面体形状和Ni-O键成键类型相对于纯TiO2都发生了较大变化,很难通过对比两者的键长、键布居等方式来确定此时Ni离子的价态。我们会在下文中,通过对电子轨道图像的分析来确定晶隙掺杂时Ni离子的价态问题。
分析数据还可以发现,相同掺杂方式的键长和键布居值随浓度变化不大,这说明在此浓度范围内的Ni掺杂对晶格的影响都较局域,掺杂的Ni原子之间不会产生明显的相互作用。
2.2 形成能分析
为了比较不同掺杂体系的稳定性,对体系的形成能进行了计算,所得形成能列于表2。形成能计算公式为[24]:

式中Eform为掺杂体系形成能,Edope、Epure分别为Ni掺杂和纯TiO2的总能量。μNi、μTi分别为Ni和Ti的化学势。化学势的具体数值和体系的生长环境有关。在贫氧环境下(O-poor),通过计算对应的金属块体能量得到μNi、μTi。富氧情况下(O-rich),通过式(2)、(3)得到μNi、μTi。式中μO为氧气中氧原子的化学势μO= 1/2μ(O2)

从表2中数据看出:贫氧环境下,Ni更倾向于进入TiO2的晶格间隙之中,而形成Niin掺杂;而富氧环境有利于Ni替Ti位掺杂NiTi的形成。另外,NiTi的形成能受氧环境的影响很大,尤其是在富氧环境下,NiTi拥有最低的形成能(-0.7162eV,-0.7127eV),说明此时NiTi很容易形成,并且稳定性很高[24-25]。在不同氧环境下,两种掺杂方式的形成能都随掺杂浓度的升高而增加,但是变化幅度不大。形成能的计算结果说明,通过控制体系形成过程中的氧环境,可以对掺杂方式加以影响,这对于实验过程中的优化是有利的。
2.3 电子结构分析
2.3.1 能带分析
为讨论Ni掺杂对体系光学性能的影响,对不同掺杂体系的能带结构进行了计算。计算结果表明,如图2(a):作为典型的半导体材料,本征TiO2的费米能级位于价带顶端。计算所得的禁带宽度为2.445eV。Ni替位掺杂NiTi时,如图2(b)(c):费米能级同样位于价带顶部,其禁带宽度Eg略大于本征TiO2,且Eg随掺杂浓度的升高而略有增大。由Ni3d-O2p轨道杂化形成的两条杂质能级位于距离价带顶0.94~1.37eV的禁带之中。这一方面说明Ni3d轨道的能量低于Ti3d轨道;另一方面也说明,Ni掺杂对晶格的扰动使配位O原子发生了能级分裂。杂质的能级位置离价带顶和导带底都较远且位于费米能级之上,价带电子和已经到达导带的电子都会优先激发或回落到杂质能级之上,这虽然有利于吸收光谱的红移,但也很容易成为光生电子-空穴的复合中心[9]。
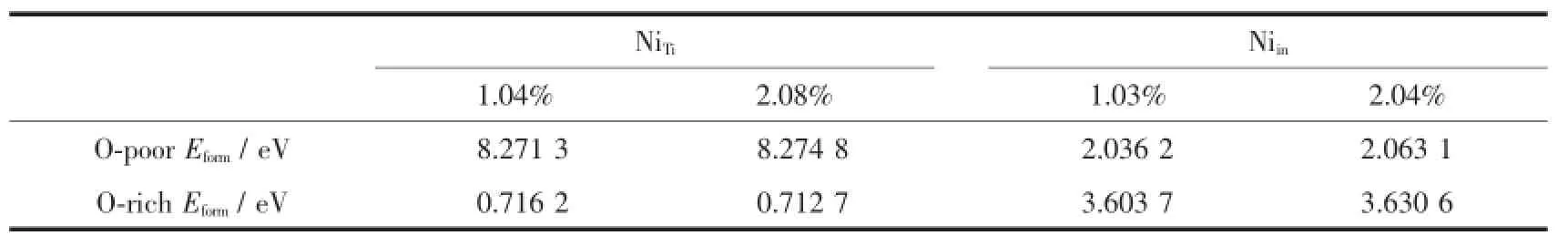
表2 掺杂体系形成能Table 2Formation energies of the doped system
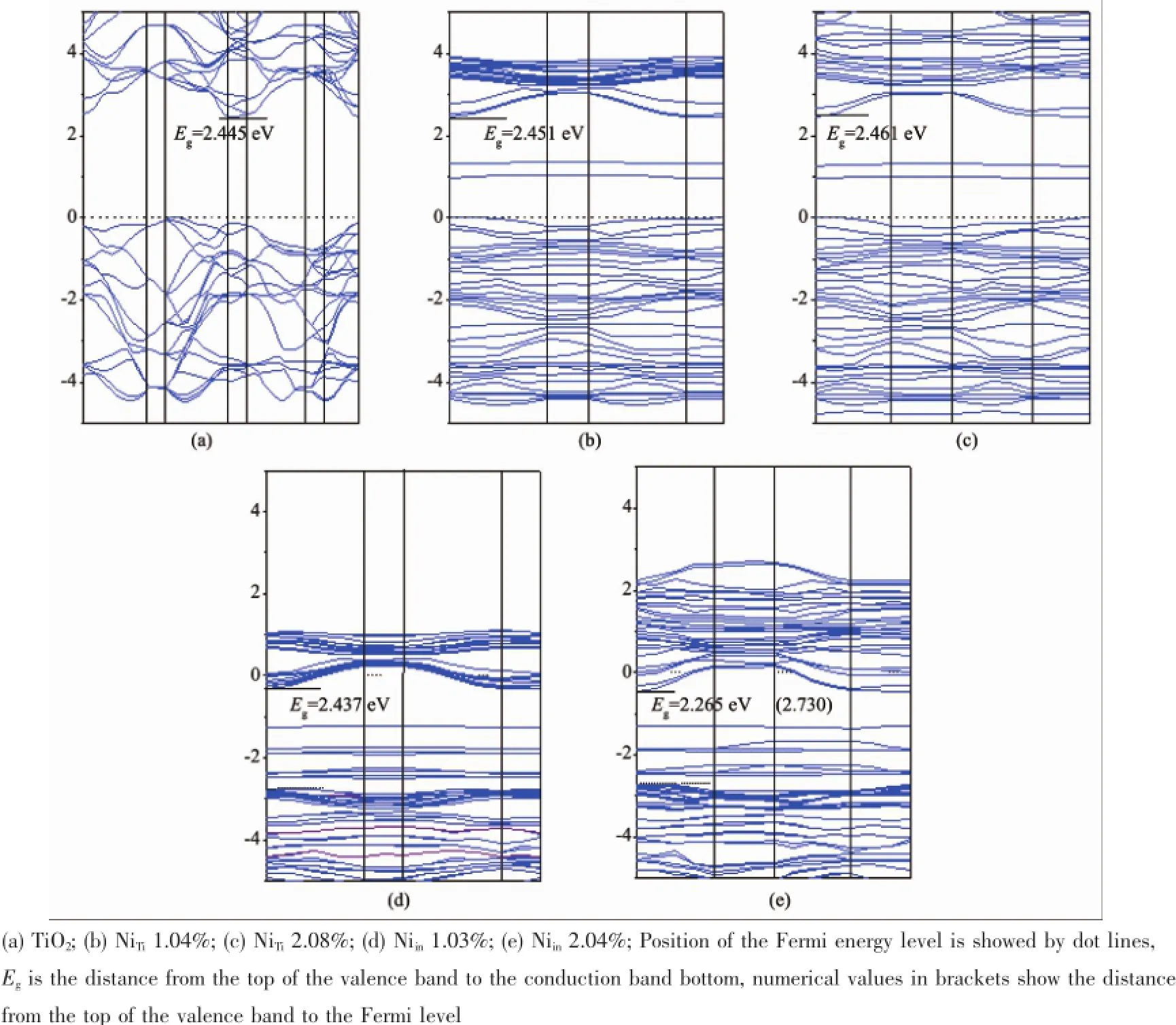
图2 纯TiO2和Ni掺杂体系能带结构图Fig.2Band structure of TiO2and Ni doping system
Ni晶隙掺杂时,如图2(d)(e):其禁带宽度相对本征TiO2减小。Niin带来的多余电子推动费米能级升高并穿过导带、载流子发生n型简并,体系呈现明显的金属性。杂质能级位于价带顶0.26~1.43eV的能量区间内且条数较多。虽然在禁带中出现杂质能级,但是由于其都位于费米能级之下,根据能带填充机理,这时电子从价带顶到最低非占据态(费米能级)的跃迁才是主要的[7],完成跃迁所需要吸收的光子能量会更大。所以,Niin情况下,吸收带边实际会发生蓝移。Niin情况下的部分电子进入位于导带的空Ti3d能级,导致Ti3+离子产生,过程可表示为Ti4++ e-=Ti3+。实验和理论计算都证明,Ti3+可以成为高效的光生电子-空穴分离中心和吸附物活化中心,并且电子在导带Ti3d态的有效质量相对较小[26-27],这都将对晶体催化性能和电学性能的提高有重要影响。在晶格间隙掺杂Ni时,导带和价带都明显向低能方向移动。研究表明,价带电位的降低有利于光生空穴的氧化性;而导带电位的升高有利于光生电子的还原能力。导带的向下移动将严重限制材料的部分应用[9]。比如,在光防腐应用中,为了使进入导带的光生电子能够顺利流入被保护的阴极金属,导带电子的电位(E/V vs NHE)必须高于金属[4]。
2.3.2 态密度和轨道分析
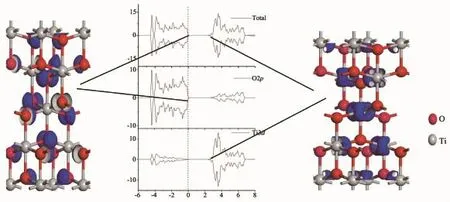
图3 本征TiO2态密度和部分能级轨道图Fig.3Density of States and the partial level track diagram of TiO2
在几何结构分析和能带分析的基础上,为了更加清晰的表明掺杂离子的价态、成键和体系的磁性情况,进行了态密度和能级轨道的研究。为便于进行比较,并使轨道图像具有较高的辨识度,所绘轨道均为20nm-3等值面。图3分别为纯TiO2态密度、价带顶能级(-0.748~0eV)和导带底能级(2.445~3.388eV)的轨道图像。可以发现,纯TiO2的价带主要由O2p轨道构成,而导带主要由Ti3d轨道构成;价带顶能级轨道只局域于O离子周围且和Ni-O键垂直。导带底能级轨道同样只局域于Ti离子周围,且指向NiO6八面体xy平面的O原子间隙方向。Asahi等认为,TiO2价带顶和导带底应该分别由非键态的O2pπ和Ti3dxy轨道构成[29]。本文的态密度和轨道分布图像都很好的符合了上述的观点。
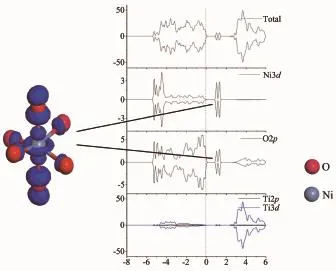
图4 NiTi掺杂态密度和部分能级轨道图Fig.4Density of States and the partial level track diagram of NiTi-doping anatase
由于在几何结构分析中发现,不同掺杂浓度不会使成键和配位场的分布有较大的改变,所以浓度的改变不会影响Ni离子的价态。本文以高浓度掺杂情况下(Niin:2.04%和NiTi:2.08%)的态密度和轨道能级图像作为研究对象。从图4可以清楚的发现替位掺杂Ni离子的3d轨道分裂情况。相对于TiO6八面体,由于配位氧原子向中心Ni弛豫,加强的晶体场使Ni3d的eg和t2g两组能级分裂更大,其中eg为空轨道,t2g为完全占据轨道。在禁带中,距离价带顶(0.94~1.37eV)的能量范围内出现由Nieg轨道和O2p轨道杂化产生的反键态杂质能级。Nit2g和O2p轨道在价带内部也形成较强的态密度峰共振,说明Ni-O在此能量范围成键。此时Ni离子为Ni4+电子态为d6(t2g3↑,t2g3↓)。这和几何分析中对Ni离子价位的判断一致。由于替位掺杂Ni离子与Ti有相同的价态,体系不需要进行电子代偿,Ni周围原子没有发生极化。此时总态密度和各原子的分波态密度图上下对称,电子自旋净磁矩(2×Integrated Spin Density=0.215960E-06)和电子自旋密度的模(2× Integrated|Spin Density|=0.237192E-03)都近似为零,表明体系为顺磁性[28]。
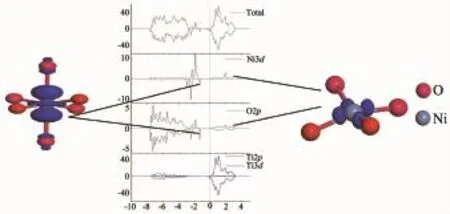
图5 Niin掺杂态密度和部分能级轨道图Fig.5Density of States and the partial level track diagra of Niin-doping anatase
Ni形成替位掺杂时,Ni3d能级的多数态密度峰在费米能级之下同O2p能级共振成键,并形成多个局域性较强的共价性杂质能级。这些杂质能级应归因于Ni和O的成键电子。分析图发现,Ni3d能级在费米能级之上(1.9eV)出现了一个较弱的空态密度峰。根据轨道能级图像,此轨道主要局域于Ni离子附近并指向NiO6八面体xy平面的4个O原子,具有明显的轨道特征。同样,通过分析杂质能级高能部分的轨道图像,发现此能级轨道为计算结果和晶体场理论都表明,此时能量高的3d为空轨道,低能量的全部为占据态。分析图可知,此时的Ni离子为Ni3+,电子态为d7精细分析发现,费米能级处的自旋向上和向下态密度图并不对称,说明晶隙掺杂的Ni离子在体系中引入的多余电子,扭曲了晶格的磁场分布,使Ni离子周围的O和Ti都发生了自旋分裂。体系具有磁性。此时的电子自旋净磁矩(2×Integrated Spin Density=1.79596)小于电子自旋密度的模(2×Integrated|Spin Density|=2.66133),说明体系具有亚铁磁性。Chen等通过研究Ni和氧空位的关系发现,Ni3+中的未成对电子是体系具有磁性的来源,只有体系存在Ni3+才会具有磁性[16]。Cho等在实验中也得到了相同的结论[17]。本文的计算结果与上述观点符合。Ni原子、Ni3+和Ni4+的3d、4s轨道电子分布可表示为图6。
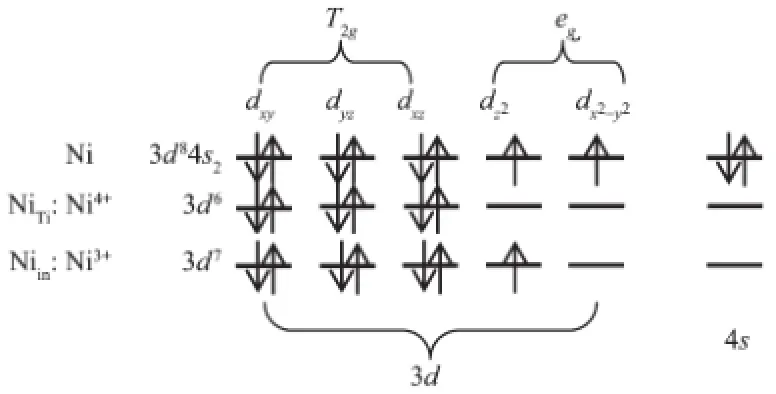
图6 Ni原子、Ni3+和Ni4+的3d、4s轨道电子分布Fig.6Distribution map of 3d,4s orbital electron of Ni,Ni3+and Ni4+
3 结论
本文通过第一性原理方法配合晶体场理论,对不同浓度NiTi(1.04%、2.08%)和Niin(1.03%、2.04%)掺杂锐钛矿TiO2的几何结构和电子结构进行了研究。计算结果表明:
(1)不同掺杂方式的Ni离子价态不同,分别为Ni4+电子态d6↑,↓:NiTi)和Ni3+电子态d7(↑,dxy↑↓,dxz↑↓dyz↑↓:Niin)。NiTi时,体系具有顺磁性;而在Niin情况下,体系在d轨道未成对电子作用下具有亚铁磁性。
(2)富氧环境下,Ni更倾向于进入TiO2的晶格间隙之中;而贫氧环境有利于Ni替位掺杂NiTi的形成。另外,NiTi的形成能受氧环境的影响很大,尤其是在富氧环境下,NiTi拥有最低的形成能(-0.7162eV,-0.7127eV),说明此时NiTi很容易形成,并且稳定性很高。
(3)NiTi掺杂时,Ni-O键长小于被取代的Ti-O键。此时的Ni-O键平均布居值小于被取代的Ti-O键,说明成键共价性减弱,这会导致晶体稳定性降低。且掺杂浓度越高,折合晶胞体积越小。Niin的过程中会向体系引入过剩的电子,晶体库伦排斥作用增强,晶胞体积增加,这也会降低晶体的稳定性。
(4)NiTi使带隙增加,而Niin使带隙减小。其改变幅度都随浓度增加而增大。两种掺杂方式的杂质能级都由Ni3d-O2p轨道杂化形成。NiTi虽然有利于吸收光谱的红移,但也很容易成为光生电子-空穴的复合中心。Niin掺杂则会导致吸收光谱蓝移,并使导带和价带都明显向低能方向移动。
[1]Gombac V,Rogatis L D,Gasparotto A,et al.Chem.Phys., 2007,339:111-123
[2]Yuan J,Fujisawa R,Tsujikawa S.Zairyo-to-kankyo,1994,43: 433-440
[3]Shen G X,Chen Y C,Lin L,et al.Electrochim.Acta, 2005,50:5083-5089
[4]Sun M M,Chen Z Y,Yu J Q.Electrochim.Acta,2013, 109:13-19
[5]Zhang X X,Chen Q C,Tang J,et al.Sci.Rep.,2014,4:560-560
[6]Yu S M,Guan X M,Wong H S P.IEEE Trans.Electron Devices,2012,59:1183-1188
[7]Yang K S,Dai Y,Huang B B.Phys.Rev.B,2007,76:195201(6pages)
[8]Geng H,Yin S W,Yang X,et al.J.Phys.:Condens.Matter, 2006,18:87-96
[9]Zhao L,Park S G,Magyari-kope B,et al.Appl.Phys.Lett., 2013,102:083506(1-4)
[10]Choi W Y,Termin A,Hoffmann M R.J.Phys.Chem.,1994, 98:13669-13679
[11]ZHAO Zong-Yan(赵宗彦),LIU Qing-Ju(柳清菊),ZHANG Jin(张瑾),et al.Acta Phys.Sin.(物理学报),2007,56(11): 6592-6599
[12]Yao Z P,Jia F Z,Tian S J,et al.ACS Appl.Mater.Interfaces,2010,2:2617-2622
[13]Visinescu C M,Sanjines R,lévy F,et al.Appl.Catal,B, 2005,60:155-162
[14]Pol R,Guerrero M,Garcia-Lecina E,et al.Appl.Catal,B, 2016,181:270-278
[15]ZHANG Xiao-Chao(张小超),ZHAO Li-Jun(赵丽军),FAN Cai-Mei(樊彩梅),et al.Acta Phys.Sin.(物理学报),2012, 61:077101(1-9)
[16]Chen J,Lu G H,Cao H H,et al.Appl.Phys.Lett.,2008,93: 172504(1-3)
[17]Cho J H,Hwang T J,Joh Y G,et al.Appl.Phys.Lett.,2006, 88:092505(1-3)
[18]KneiB M,Jenderka M,Brachwitz K,et al.Appl.Phys.Lett., 2014,105:062103(1-5)
[19]Niishiro R,Kato H,Kudo A.Phys.Chem.Chem.Phys., 2005,7:2241-2245
[20]Aryasetiawan F,Karlsson K,Jepsen O,et al.Phys.Rev.B, 2006,74:5106
[21]RuizPreciado M A,Kassiba A,Morales-Acevedo A,et al. RSC Adv.,2015,5:17396-17404
[22]Yang J,Lv C Q,Guo Y,et al.J.Chem.Phys.,2012,136: 104107(1-14)
[23]Luttrell T,Halpegamage S,Tao J G,et al.Sci.Rep.,2014, 4:447-467
[24]Yu J G,Zhou P,Li Q.Phys.Chem.Chem.Phys.,2013,15: 12040-12047
[25]Segall M D,Lindan P J D,Probert M J,et al.J.Phys: Condens.Matter.,2002,14:2717-2744
[26]Fan Z,Wang L,Wu T,et al.J.Am.Chem.Soc.,2010,132: 11856-11857
[27]Cheng H Z,Selloni A.J.Chem.Phys.,2009,131:9249-9260
[28]WANG Hong-Ming(王宏明),ZHENG Rui(郑瑞),LI Gui-Rong(李桂荣),et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31(11):2143-2151
[29]Asahi R,Taga Y,Mannstadt W,et al.Phys.Rev.B,2000, 61:7459-7465
Study on Ni-Doped Anatase by First Principle Method
YU Zhi-Qing1,3WANG Xun3TIAN Ang1,2LIU Yan-Xia4,5YANG He1,2XUE Xiang-Xin*,1,2
(1School of Metallurgy,Northeastern University,Shenyang 110819,China)
(2Key Laboratory of Liaoning province for Recycling Science of Metallurgical Resources,Shenyang 110819,China)
(3School of Science,Shenyang Jianzhu University,Shenyang 110168,China)
(4School of Physics,Liaoning University,Shenyang 110036,China)
(5Titanium Alloys Division,Institute of Metal Research,Chinese Academy of Sciences,Shenyang 110016,China)
The geometric structures,formation energies,electronic structures and magnetic properties of Ni-doped anatase,with different concentrations and doping methods,were studied by the first principle method under the framework of the spin density functional theory,combined with the crystal field theory.The results of formation energy calculation show that the oxygen environment,in the process of crystal growth,has an important influence on the structures of Ni-doping.Analysis of the state density and energy level track map showed that the valence states of Ni ion was different in different doping conditions.Impurity energy levels in the bandgap of all kinds of doping system are formed by the hybridization of Ni3d-O2p.When a doping Ni ion substitute lattice Ti,that will make the unit cell volume and the crystal stability decrease,absorption spectrum red shift,the system is paramagnetic.Interstitial Ni doping makes the unit cell volume increase,the absorption spectrum blue shift,and enables the band to move in the direction of low energy.At this time,the carrier occur n type-degeneration,theability,of some Ti ions,of losing electrons declined,resulting in the making of Ti3+ions,the system has magnetism.
Ni-doping;the first principles method;electronic structures;formation energy
O614.81+3
A
1001-4861(2017)01-0041-08
10.11862/CJIC.2017.015
2016-04-01。收修改稿日期:2016-10-22。
国家自然科学基金(No.U1261120)资助项目。
*通信联系人。E-mail:xuexx@mail.neu.edu.cn
