分步预硫化Mo-Ni/γ-Al2O3催化剂的制备、表征及加氢催化性能
2017-02-08王鼎聪赵德智杨辰思张志伟
张 强, 丁 巍,3, 王鼎聪, 赵德智, 杨辰思, 张志伟
(1.辽宁石油化工大学 化学化工与环境学部, 辽宁 抚顺 113001; 2.中国石化 抚顺石油化工研究院, 辽宁 抚顺 113001;3.中国石油大学 重质油国家重点实验室, 北京 102249)
分步预硫化Mo-Ni/γ-Al2O3催化剂的制备、表征及加氢催化性能
张 强1, 丁 巍1,3, 王鼎聪2, 赵德智1, 杨辰思1, 张志伟1
(1.辽宁石油化工大学 化学化工与环境学部, 辽宁 抚顺 113001; 2.中国石化 抚顺石油化工研究院, 辽宁 抚顺 113001;3.中国石油大学 重质油国家重点实验室, 北京 102249)
采用自组装法将硫脲、柠檬酸及聚乙二醇-200制成器外预硫化沉淀液,原位沉淀法制备了分步预硫化Mo-Ni/γ-Al2O3催化剂。采用BET、XRD、CO原位吸附、XPS、TEM等手段表征所制备的催化剂,采用固定床微型反应器评价催化剂的加氢催化性能,并与直接器内预硫化法制备的催化剂对比。结果表明,分步预硫化法制备的催化剂在第一次器外预硫化后硫化度达到74.8%;聚乙二醇-200和硫脲的加入可以改善催化剂孔性质,且当硫脲含量达到20%(占Mo、Ni金属氧化物质量和)时,催化剂的比表面积达到198 cm2/g,此时,其活性金属负载分散性好,硫化度比对比剂提高了2.60百分点。分步预硫化Mo-Ni/γ-Al2O3催化剂催化FCC柴油加氢反应40 h的加氢脱硫、脱氮率及芳烃饱和率比对比剂分别高出2.28百分点、1.94百分点及3.46百分点。
分步预硫化; 原位沉淀; 催化剂; 加氢脱硫(HDS); 加氢脱氮(HDN)
面对现今石油能源环境,重油和渣油加氢技术已经成为主要的研究方向[1-3],催化剂更是其中的重点。载体性质、活性金属负载方式等直接影响加氢催化剂的活性。载体是催化剂的关键组成部分,对催化剂的孔结构、表面酸性、电性质等都有很大影响[4]。活性氧化铝作为催化剂载体效果显著[5-6]。王鼎聪[7]采用纳米自组装方法合成了大孔容介孔氧化铝,田野等[8]以此为载体制备的催化剂在劣质油加氢中表现出了良好的活性及稳定性,鄢景森等[9]研究的TiO2-Al2O3载体表现出了良好的孔结构性质及较高的金属负载分散性。
加氢催化剂的活性组分Mo、Ni、Co、W等元素一般以金属氧化物形式存在,在进行加氢反应前要进行预硫化处理,以提高催化剂的活性和选择性[10-13]。人们对预硫化机理已经进行了广泛的研究[14],器内预硫化技术已很成熟,器外预硫化技术也有很多研究报道。GAO等[15]详细研究并比较了器內及器外预硫化技术的特点,夏远亮[16]采用原位分解法制备了免预硫化CoMo/γ-Al2O3催化剂,林凌等[17]制备的免预硫化加氢脱硫MoNiP/Al2O3催化剂拥有较高的催化活性,田维乾等[18]制备的器外预硫化催化剂表现出较高的催化特性。但器外预硫化技术由于预硫化液黏度大等原因,实现工业化和大规模生产有诸多困难[19]。
笔者将硫脲及聚乙二醇-200直接加入到活性金属浸渍液中,浸渍后置于微型反应器中进行原位沉淀反应,制备一种免焙烧、分步预硫化的加氢催化剂。采用BET、CO原位吸附、XRD、XPS等手段及催化剂加氢实验表征和评价所制备的催化剂,并与器内预硫化法制备的催化剂对比,为制备免焙烧、分步预硫化催化剂提供参考。
1 实验部分
1.1 试剂
MoO3,纯度大于99.50%;碱式碳酸镍,Ni质量分数高于44.00%;磷酸,w(H3PO4)≥85%;柠檬酸,纯度大于99%;硫脲,分析纯;聚乙二醇-200(PEG-200),分析纯,沈阳市新化试剂厂产品。3%预硫化液(CS2体积分数为3%的航空煤油),自制。催化裂化柴油,中国石化镇海炼化分公司提供。
1.2 催化剂的制备
取MoO3160.4 g、碱式碳酸镍45.9 g、磷酸59.3 g及去离子水200 mL放入烧瓶中,置于山东鄄城华鲁电热仪器有限公司SHT数量调温搅拌电热套中,在设置温度170℃、溶液实际温度100℃下反应3 h。过滤,向滤液中加入70%(占Mo、Ni金属氧化物质量和)柠檬酸,充分搅拌使其溶解。将溶液平均分成两份,一份直接定容250 mL,得到Mo/Ni质量比为6的金属浸渍液A;另一份加入40%(占Mo、Ni金属氧化物质量和)PEG-200后,定容250 mL得到溶液B。
取溶液A加入5%(占Mo、Ni金属氧化物质量和)的硫脲,再取溶液B三份分别加入0、5%和20%的硫脲。将上述4种溶液分别饱和浸渍60 mL载体,浸渍后载体编号为1、2、3、4号。将2号载体110℃烘干3 h,程序升温至200℃焙烧2 h,400℃焙烧4 h制得催化剂MNAP401;1、3、4号3个样品置于沈阳施博达仪器仪表有限公司微型水热反应釜中,在150℃下进行原位沉淀反应6 h,取出后在110℃下烘干3 h,分别制得器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20。
取催化剂MNAP401和MNAP40-20,以3%CS2(质量分数)的航空煤油为硫化剂,在反应压力6 MPa、氢/油体积比600、空速3.2 h-1条件下,程序升温在90、170、240℃分别停留3 h,370℃停留10 h,进行器内预硫化,制得器内预硫化催化剂MNAP40和分步预硫化催化剂MNAP40-202。
1.3 器外预硫化原位沉淀反应机理
当硫脲作为沉淀剂,在水热反应中,硫脲中的S充当了反应的还原剂,原料中的S被氧化,生成SO42-;同时,它又是优良的硫化剂,为反应提供了硫源,生成MoS2。实际上,硫脲[CS(NH2)2]和水反应可分解为CO2、H2S和NH3[20],如式(1)所示,生成的H2S还原性很强,可以将MoO42-中的Mo6+还原为Mo4+,可能的反应如式(2)所示。
2NH3(g)+H2S(g)+CO2(g)
(1)
4MoS2+SO42-+6SCN-+24NH3(g)+9CO2(g)
(2)
在催化剂XRD表征中检测出N2H6SO3的特征衍射峰,推测为反应中生成的副产物,可能的反应如式(3)所示。
N2H6SO3+2SCN-+3H2O
(3)
此外,硫脲分解生成的NH3和CO2少部分溶于水得 (NH4)2CO3,大部分以气体形式离开了反应体系。
1.4 催化剂的活性评价
采用沈阳施博达仪器仪表有限公司连续进料的固定床微型反应器评价催化剂的加氢催化性能。催化剂装填量20 mL,在压力6 MPa、温度370℃、氢/油体积比600、液体质量空速1.6 h-1的条件下,对劣质催化裂化柴油进行加氢。稳定10 h后开始取样,之后每隔4 h取一次样,直到40 h结束。原料催化裂化柴油的性质见表1。

表1 加氢实验用催化裂化柴油的性质Table 1 Properties of the FCC diesel fraction for hydrotreating
1.5 催化剂的表征方法
采用美国Micromeritics公司ASAP 2420型物理吸附仪测量催化剂孔性质,样品先在90℃下脱气30 min,再200℃下脱气2 h,以N2作为吸附质,在液氮(-196℃)下进行测定。采用日本理学公司 D/2500 型X-射线衍射仪测定样品晶相结构(XRD),光源CuKα,波长0.154 nm,管工作电压40 kV,工作电流80 mA。采用Thermo公司Nicolet 6700型傅里叶变换红外光谱仪测定催化剂的CO原位吸附红外光谱,常压,500℃,高纯氢气还原3 h,300℃高真空净化,室温吸附CO,向原位池中引入少量CO气体,吸附平衡30 min后,脱附至0.1 mPa。采用美国Perkin-Elmer Physics Electronics公司PIH 5300 X-射线光电子能谱仪获取催化剂的XPS谱,MgKaX射线单色化激发源,功率150 W,荷电效应用来自载体Al2O3的Al(2p)峰(74.7 eV)校正。采用日本电子光学公司JEM-2000 型高分辨透射电镜进行TEM分析,实际放大倍数30万倍,加速电压200 kV,LaB6灯丝,线分辨率0.14,点分辨率0.23 nm。催化剂样品均保存于乙醇溶液中以避免与空气接触,每次测量时取少量催化剂于玛瑙研钵中研细后超声波分散于乙醇溶液中,取少量的悬浮液置于涂炭铜筛网上制样,进行分析。
2 结果与讨论
2.1 PEG-200及硫脲用量对器外预硫化催化剂性质的影响
考察了PEG-200及硫脲的加入量对器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20的孔性质和金属负载量的影响,结果列于表2。
从表2可见,与MNAL-5相比较,MNAP40-5的最可几孔径有所减小,但比表面积和孔容分别增大了57 m2/g和0.11 m3/g,且活性金属负载量增加,说明PEG-200有扩孔及改善金属分散性的作用;MNAP40-20孔容增加了0.07 cm3/g,比表面积增加了43 m2/g,说明硫脲的加入,一方面可以提供预硫化的硫来源,另一方面,可以对催化剂起到扩孔的作用。
图1为器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20的最可几孔径。由图1可以看出,3种催化剂均出现了双介孔结构,既存在提供加氢反应活性位的小孔,也存在为大分子反应物和产物提供扩散作用的大孔,这种等级介孔的存在有助于提高催化剂的加氢反应催化活性。
图2为器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20的N2吸附-脱附等温线。由图2可以看出,3种催化剂的N2吸附-脱附等温线均属于类型Ⅳ[21],都具有在较低相对压力(p/p0)区曲线平稳向上、在较高p/p0区等温线迅速上升的特点。从滞后环的形状来看, MNAL-5 的大部分吸附存在于p/p0大于0.8的高压区,说明此催化剂的孔道均以较大介孔为主,但吸附量较其他两个催化剂少;MNAP40-20和MNAP40-5滞后环在p/p0为0.45处就开始迅速上升,一直持续到p/p0为1.0时,说明其孔道均属于介孔范围,且活性金属在催化剂表面以多层吸附状态存在。
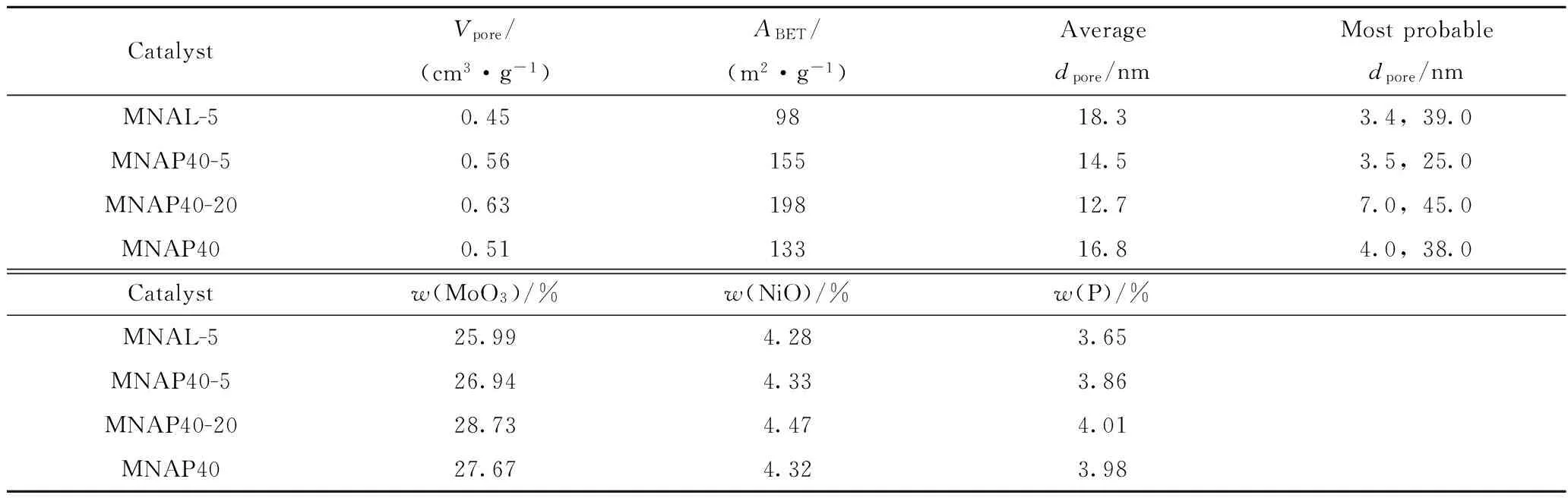
表2 器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20的孔结构性质和金属负载量Table 2 The texture properties and metal loadings of MNAL-5,MNAP40-5 and MNAP40-20 catalysts
Vpore—Pore volume;ABET—Specific surface area;dpore—Pore diameter.
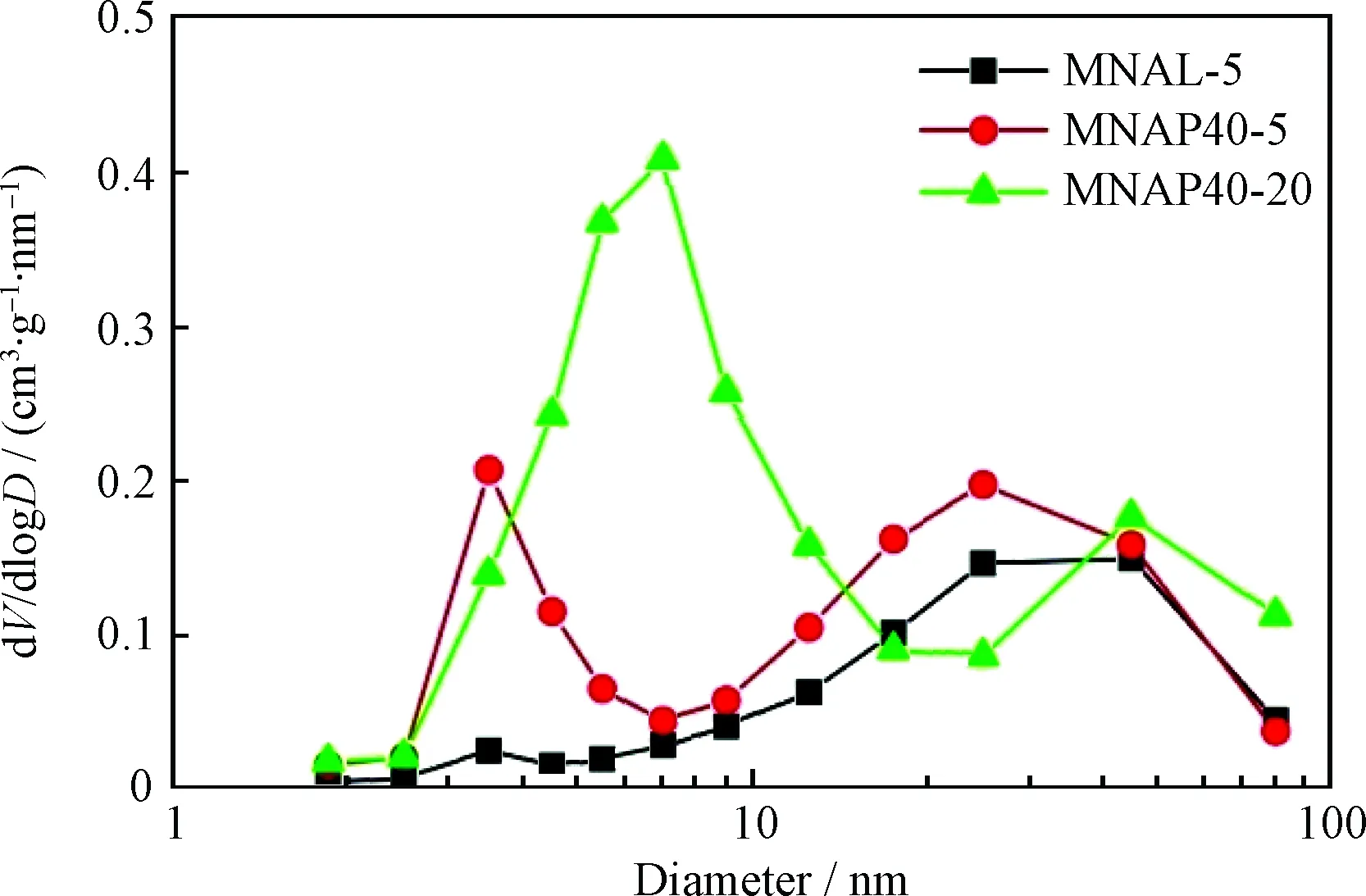
图1 器外预硫化催化剂MNAL-5、MNAP40-5和 MNAP40-20的最可几孔径Fig.1 Most probable pore of MNAL-5, MNAP40-5 and MNAP40-20 catalysts
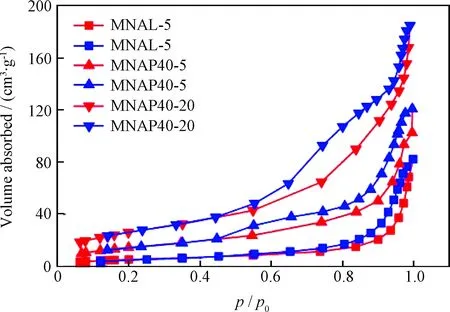
图2 器外预硫化催化剂MNAL-5、MNAP40-5和 MNAP40-20的N2吸附-脱附等温线Fig.2 N2 adsorption-desorption isothermals of MNAL-5, MNAP40-5 and MNAP40-20 catalystsAdsorption curve; Desorption curve
2.2 所制备的Mo-Ni/γ-Al2O3催化剂的表征结果
1.创新管理机制。针对当前社会组织的管理现状,必须坚持油田党委和社会组织党组织负责日常管理,业务主管部门、行业协会加强协调联系的管理模式,改变过去没有婆家或者“谁才是婆家”管理主体不明晰的局面,明确工作责任,落实工作职责,形成覆盖全区、上下贯通的社会组织党建工作管理体系。
2.2.1 XRD分析
图3为MNAL-5、MNAP40-5和MNAP40-20催化剂的XRD谱。从图3可见,在2θ为37.44°、45.78°和67.31°处(PDF#04-080)明显出现了γ-Al2O3的(311)、(400)和(441)晶面的特征衍射峰,在2θ为14.53°、33.21°、39.5°和58.32°处(PDF#17-0744)出现了MoS2的(003)、(101)、(103)和(110)晶面的特征衍射峰,且随着PEG-200的加入及硫脲含量的增加,它们的特征衍射峰越来越明显,说明活性Mo4+的量越来越多;尤其是在2θ为14.53°和58.32°处MNAP40-20的MoS2特征衍射峰最强,且峰型弥散,说明此催化剂MoS2的含量高,并在载体的内、外表面分散均匀。
MNAL-5的XRD谱中,在2θ为36.52°和53.79°处(PDF#50-0739)出现了MoO2的(100)和(102)晶面的特征衍射峰,在2θ为23.01°和25.00°处(PDF#47-1081)出现了MoO3的(011)和(200)晶面的特征衍射峰,说明此催化剂中还存在氧化态Mo6+及Mo4+。此种现象是由于器外预硫化不充分导致。MNAP40-5和MNAP40-20样品的这些衍射峰趋于消失,证明随着PEG-200的加入和硫脲含量的增加,样品中有更多的氧化态钼转变成硫化态钼,催化加氢反应活性位增加;MNAP40-20在2θ为32.21°和48.84°处(PDF#12-0041)出现了NiS的(300)和(131)晶面的特征衍射峰,且随着PEG-200的加入和硫脲含量的增加,这些特征衍射峰趋于弥散,说明具有更良好的金属分散性。此外,MNAL-5在2θ为20.34°处(PDF#42-0659)出现了N2H6SO3的(112)晶面的特征衍射峰,但是MNAP40-5和MNAP40-20的此峰明显减小甚至消失,与此同时Mo4+的特征衍射峰明显增强,说明PEG-200的加入可以减少副产物的生成。
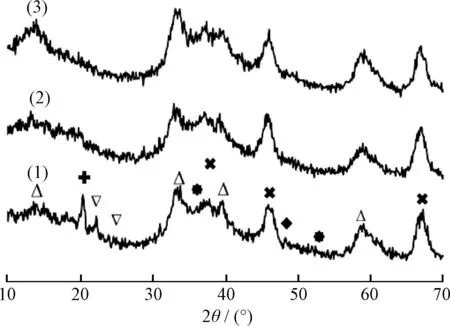
图3 器外预硫化催化剂MNAL-5、MNAP40-5和 MNAP40-20催化剂的XRD谱Fig.3 XRD patterns of MNAL-5,MNAP40-5 and MNAP40-20 catalysts(1) MNAL-5; (2) MNAP40-5; (3) MNAP40-20 MoS2; MoS3; NiS; MoS2; γ-Al2O3; N2H6SO3
2.2.2 CO原位吸附红外光谱
CO作为探针的红外光谱被广泛应用于催化剂表面活性金属及其存在状态的表征[22]。器外预硫化催化剂MNAL-5、MNAP40-5和MNAP40-20的CO原位吸收红外光谱示于图4。
图4显示,3种催化剂均产生了<2000 cm-1及>2000 cm-1的CO特征吸收峰,说明CO既有在Ni/Al2O3上的桥式吸附态[23],也有在Mo离子(Mo4+、Mo5+、Mo6+)上的线式吸附态。2187 cm-1处为CO在Mo4+上(Mo4+-CO)的吸附峰,2152 cm-1为CO在Mo5+上(Mo5+-CO)的吸附峰,2084 cm-1处接近于CO在纯Ni硫化相(Ni2+-CO)上的吸附峰[24],2036 cm-1为CO在Mo6+上(Mo6+-CO)的吸附峰,1967 cm-1及1922 cm-1为CO在Ni/Al2O3上的桥式吸附的吸附峰。随着PEG-200的加入及硫脲含量的增加,在2187 cm-1处CO吸附于Mo4+的峰和在2084 cm-1处CO吸附于Ni硫化相的峰均逐渐增强,而在2036 cm-1处CO吸附于Mo6+的峰和在2152 cm-1处CO吸附于Mo5+的峰均逐渐减弱,说明催化剂中有更多的Mo6+转变成活性Mo4+,也有更多的氧化态Ni转变成硫化态Ni,使催化剂活性位增加。
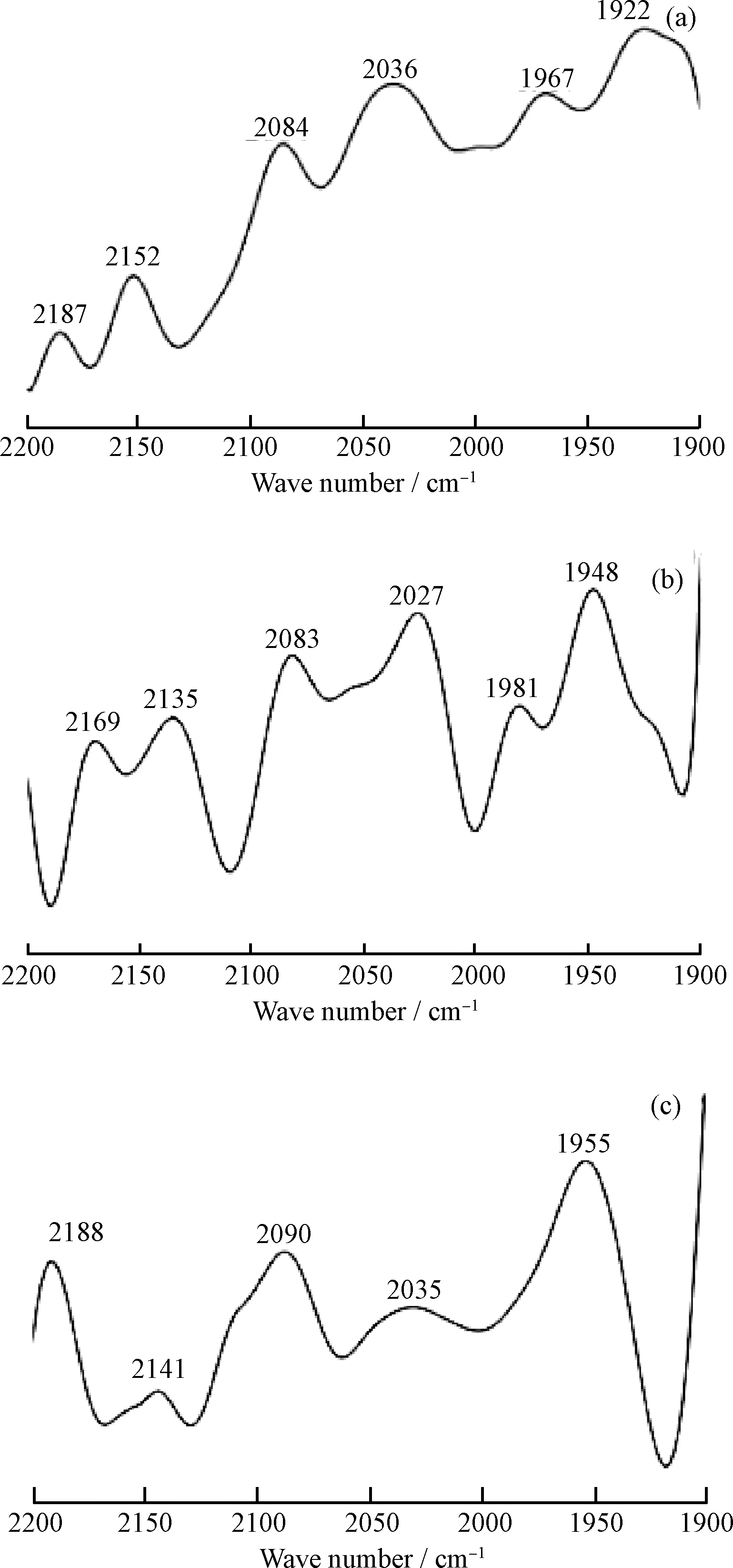
图4 MNAL-5、MNAP40-5和MNAP40-20的 CO原位吸附红外光谱Fig.4 FT-IR spectra of CO adsorbed for MNAL-5, MNAP40-5 and MNAP40-20 catalysts(a) MNAL-5; (b) MNAP40-5; (c) MNAP40-20
2.2.3 XPS表征
XPS分析能得出活性组分的种类、存在状态、在催化剂表面的分散状态及其与载体的作用方式等信息[25]。图5为硫化态催化剂MNAP40-20、MNAP40和MNAP40-202的Mo3d的XPS谱。
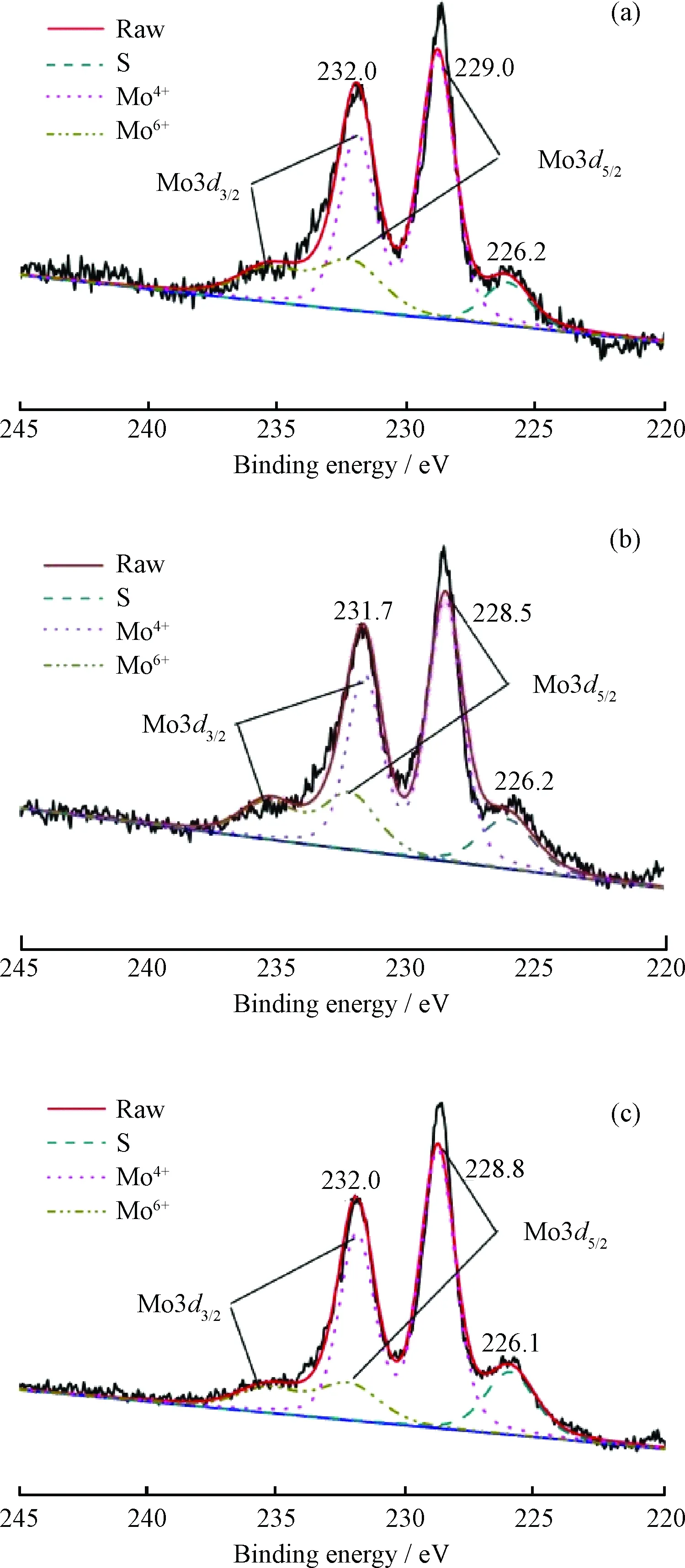
图5 硫化态催化剂MNAP40-20、MNAP40和 MNAP40-202的Mo3d XPS谱Fig.5 Mo3d XPS profiles of MNAP40-20, MNAP40 and MNAP40-202 catalysts(a) MNAP40-20; (b) MNAP40; (c) MNAP40-202
由图5可知,它们均存在2种价态的钼物种。主峰的结合能分别在232.0、231.7和232.0 eV,可归属于 +6 价的MoO3(Mo3d3/2结合能为232.5 eV)、Al2(MoO4)3(Mo3d3/2结合能为231.7 eV)或NiMoO4(Mo3d3/2结合能为232.8 eV);另一峰的结合能分别为229.0、228.5和228.8 eV可归属于+4价的MoS2(Mo3d5/2结合能为228. 9 eV)或低价钼的氧硫物种。所得硫化后主峰的结合能数值比理论值稍小,是因为硫化作用可使钼物种趋向于生成MoO2或者正六价钼的硫化形式[26]。
图6为硫化态催化剂MNAP40-20、MNAP40和MNAP40-202的Ni2p的XPS谱。结合能856.2 eV处的峰归属于与氧化铝相互作用的Ni2+物种,854.0 eV处归属于NiMoS相Ni(NiMoS)的结合能,853.2 eV处归属于Ni的硫化物。

图6 硫化态催化剂MNAP40-20、MNAP40和 MNAP40-202的Ni2p XPS谱Fig.6 Ni2p XPS profiles of MNAP40-20, MNAP40和MNAP40-202 catalysts(a) MNAP40-20; (b) MNAP40; (c) MNAP40-202
表3为XPS测得的MNAP40-20、MNAP40和MNAP40-202不同价态的Mo、Ni表面原子比和相对含量。从表3可见, MNAP40-20的Mo4+(MoS2)的峰面积占钼物种总体比例的74.8%,证明催化剂在制备过程中已完成第一步器外预硫化的过程,具有一定的活性硫化态Mo4+。MNAP40比MNAP40-20的硫化度高4%,说明在实验条件下只经过器外预硫化的硫化效果没有器内硫化的硫化效果好。但是,经过分步预硫化后,MNAP40-202催化剂的Mo4+相对含量最高,达81.4%,比直接器内预硫化催化剂MNAP40的硫化度高2.6百分点,说明采用的分步预硫化法制备的催化剂具有较高的硫化度。此外,MANP40-202有最高的NiMoS比例,分别比MNAP40及MNAP40-20
高5.7百分点及6.6百分点。边缘镶嵌Ni颗粒的MoS2形成的NiMoS相的边角具有加氢脱硫(HDS)和加氢脱氮(HDN)活性[27],说明MNAP40-202应具有最高的脱硫及脱氮催化活性。由表3还可知, MNAP40-202的Mo/Al及Ni/Al比最低。说明其表面与载体氧化铝作用的Mo及Ni的比例最低,而独立Mo、Ni硫化物的数量将随之增加[27],此催化剂应该具有最高的催化活性,这与加氢评价结果相吻合。
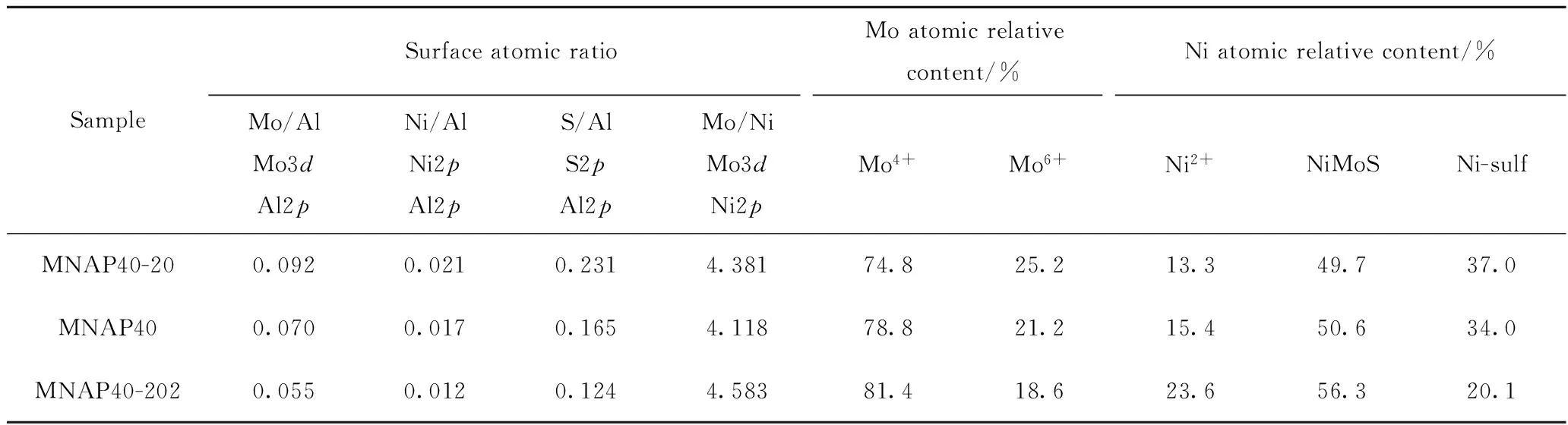
表3 MNAP40-20、MNAP40和MNAP40-202中不同价态的Mo、Ni表面原子比和相对含量Table 3 Surface atomic ratio and content of different valences Mo, Ni specie for MNAP40-20, MNAP40 and MNAP40-202 catalysts
2.2.4 TEM分析
图7为硫化态催化剂MNAP40-20、MNAP40和MNAP40-202的TEM照片。图7中,平行黑线间距为0.61 nm左右,是典型的MoS2晶面间距[28-29],多层黑线代表多层MoS2结构。3个样品均有平行黑线层,可确认活性金属在各催化剂表面均有效负载,且均为多层堆积,此现象与前面的BET表征相吻合。其中, MNAP40-20和MNAP40表面活性金属平均堆积层数分别为2~3层和3~6层,平均长度分别8~10 nm和6~8 nm,而MNAP40-202表面活性金属的堆积层数很高,甚至达到6~8层,而平均长度达8~10 nm。MoS2堆积层数的增加能够消除平躺吸附的反应物分子的空间位阻,有利于加氢反应的进行[27]。一般HDS反应发生在MoS2的角位,HDN反应发生在MoS2的边位和角位,在一定程度内高的金属堆积层数有利于HDS和HDN反应的发生,相比之下MNAP40-202应具有最高的加氢脱硫、脱氮催化活性。

图7 MNAP40-20、MNAP40和MNAP40-202的TEM照片Fig.7 TEM images of MNAP40-20, MNAP40 and MNAP40-202 catalysts(a) MNAP40-20; (b) MANP40; (c) MANP40-202
2.3 MNAP40-20、MNAP40和MNAP40-202的加氢反应催化活性
MNAP40-20、MNAP40及MNAP40-202加氢反应催化活性示于图8。
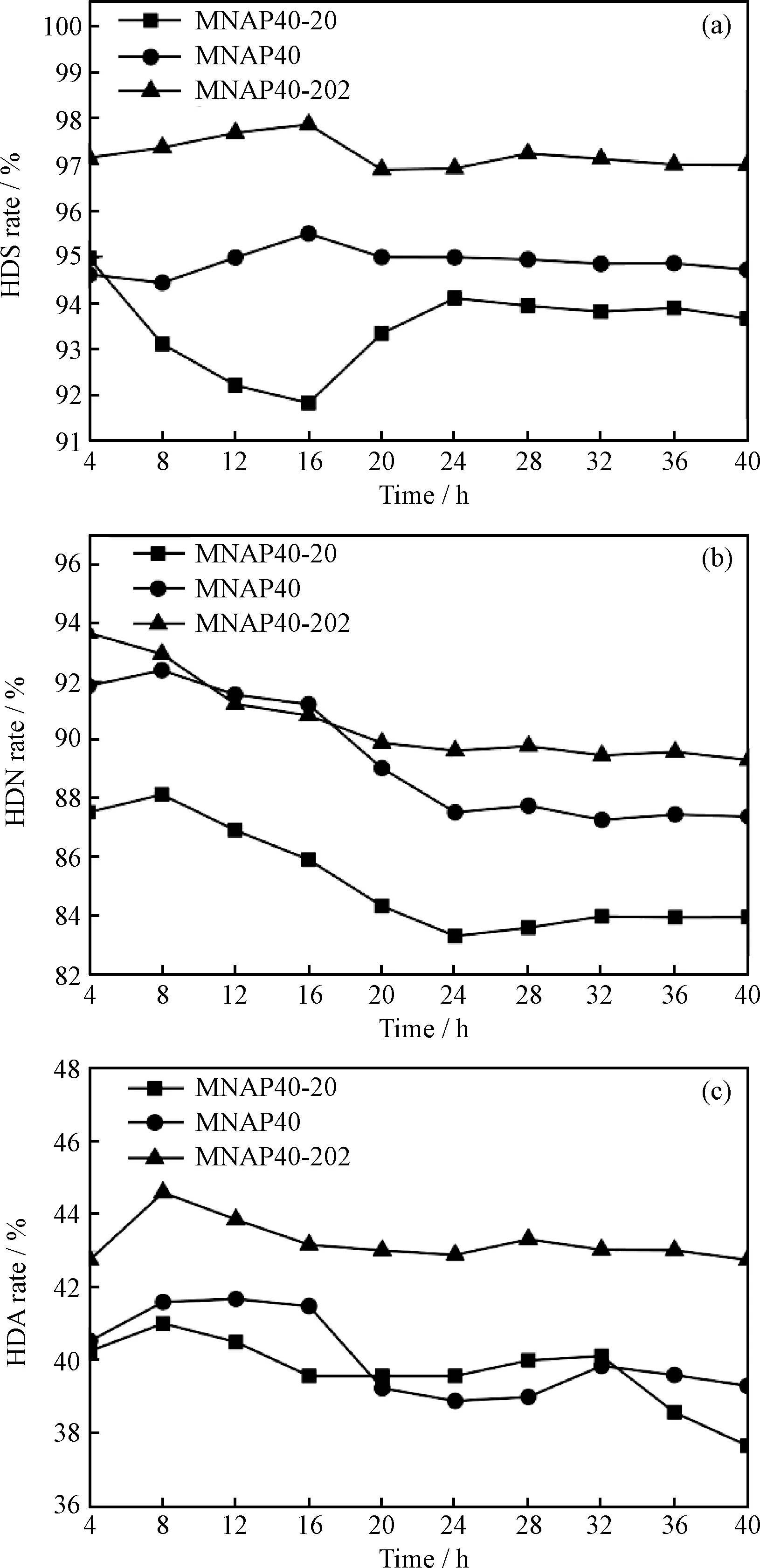
图8 MNAP40-20、MNAP40和MNAP40-202 催化加氢活性随时间的变化Fig.8 Hydrogenation performances of MNAP40-20, MNAP40 and MNAP40-202 catalysts vs reaction time(a) HDS performance; (b) HDN performance;(c) HDA performance Reaction conditions:Temperature of 370℃; Liquid hourly space velocity of 1.6 h-1; Pressure of 6 MPa
由图8(a)可看出,经3种预硫化方法处理的柴油加氢催化剂的脱硫率最终都趋于稳定,其平均脱硫活性从高到底的顺序为MNAP40-202、MNAP40、MNAP40-20,且分步硫化法制备的催化剂MNAP40-202在40 h反应结束时脱硫率稳定在97%;而只器外预硫化的催化剂MNAP40-20的脱硫率最终稳定在93.66%,其脱硫率在4~16 h出现了下降的趋势后又逐渐上升,可能由于预硫化不完全,反应中活性金属被生成的H2S对催化剂起到了一定的硫化作用造成的;器内预硫化的催化剂MNAP40的最终脱硫率稳定在94.72%。上述结果证明,分步预硫化的催化剂在柴油加氢脱硫反应中的活性高于直接器内预硫化法制备的催化剂。由图8(b)可以看出,MNAP40-202、MNAP40及MNAP40-20 3种催化剂的40 h柴油加氢脱氮率变化趋势基本相同,最终脱氮率分别稳定在89.32%、87.38%及75.93%,说明分步预硫化的催化剂加氢脱氮活性也高于直接器内预硫化处理的催化剂。如图8(c)所示,分步预硫化的催化剂MNAP40-202和直接器内预硫化的催化剂MNAP40均具有良好的芳烃饱和能力,在反应16 h后趋于稳定。MNAP40-202最终稳定的转化率达到42.75%,稍高于催化剂MNAP40的39.29%;而只器外预硫化的催化剂MNAP40-20的芳烃转化率随着时间的延长,其活性逐渐减弱,40 h后转化率为37.65%。此现象可能由于器外预硫化法的预硫化液黏度过大,在本实验条件下预硫化不完全,催化剂的初始反应活性低,大分子芳烃生成的较大有机产物覆盖催化剂表面造成孔道部分堵塞[30-31],降低了最终催化剂处理芳烃大分子的能力。
3 结 论
(1)利用原位沉淀法制备分步预硫化催化剂过程中, PEG-200以及硫脲的加入可以改善此系列催化剂的孔结构。当硫脲加入量为20%时,MNAP40-20的孔性质最好,其孔容为0.63 cm3/g、最可几孔径为7.0和45.0 nm、比表面积为198 cm2/g,并且具有较好的金属分散性。
(2)原位沉淀法制备分步预硫化催化剂在制备过程中已有74.8%的Mo被硫化,此时催化剂已经具有催化活性,但比器内预硫化催化剂的硫化度低,而经分步预硫化后催化剂硫化度高达81.4%。
(3)催化加氢反应40 h后稳定,原位沉淀法制备的分步预硫化催化剂催化加氢所得脱硫率、脱氮率和芳烃饱和率分别达到97.00%、89.32%和42.75%,比直接器内预硫化催化剂催化所得的分别增加了2.28百分点、1.94百分点和3.46百分点,说明此方法处理的催化剂具有较高的加氢催化性能及稳定性,为制备免焙烧、分步预硫化催化剂的方法提供一定的科学依据。
[1] LIU Y D, GAO L, WEN L Y. Recent advance in heavy oil hydroprocessing technologies[J].Recent Patents on Chemical Engineering, 2009, 2(1): 22-36.
[2] MOHAN S R, JORGE A, DIAZ J A. A review of recent advances on process technologies for upgrading of heavy oils and residua[J].Fuel, 2007, 86(9): 1216-1231.
[3] SUNHWAN H, JOONGWON L, JEONG G S. Pd catalyst supported on SiO2-A12O3xero gel for hydrocracking of paraffin wax to middle distillate[J].Journal of Industrial & Engineering Chemistry, 2011, 17(2): 310-315.
[4] JOONGWON L, SUNHWAN H, SANG B L, et al. Production of middle distillate through hydrocracking of paraffin wax over NiMo/TiO2-SiO2catalysts[J].The Korean Journal of Chemical Engineering, 2010, 27(6): 1755-1759.
[5] 张志民, 郭长友, 凌凤香, 等. 氧化铝表面钛改性的机理分析[J].石油炼制与化工, 2012, 43(10): 49-54. (ZHANG Zhimin, GUO Changyou, LING Fengxiang, et al. Mechanism analysis of Ti-modifide alumina support[J].Petroleum Processing and Petrochemicals, 2012, 43(10): 49-54.)
[6] SUNHWAN H, JOONGWON L, SUNYANG P. Production of middle distillate through hydrocracking of paraffin wax over NiMo/SiO2-A12O3catalysts: Effect of SiO2-A12O3composition on acid property and catalytic performance of NiMo/SiO2-AI2O3catalysts[J].Catalysis Letters, 2009, 129(1/2): 163-169.
[7] 王鼎聪. 纳米自组装合成大孔容介孔氧化铝[J] .中国科学, 2009, 39(5): 420-431. (WANG Dingcong. Mesoporous aluminum oxide support with large pore volume by nano self-assembly[J].Science in China, 2009, 39(5): 420-431.)
[8] 田野, 赵德智, 王鼎聪, 等. 自组装催化剂在混合油加氢精制中的应用[J].石油化工高等学校学报, 2014, 27(2):15-20. (TIAN Ye, ZHAO Dezhi, WANG Dingcong, et al. The application of self-assembly catalyst in hydrorefining performance for mixed oil[J].Journal of Petrochemical Universities, 2014, 27(2): 15-20.)
[9] 鄢景森, 王海彦, 张静茹, 等. TiO2-Al2O3载体的制备方法对其负载的磷化镍催化剂加氢脱氮反应性能的影响[J].物理化学学报, 2014, 30(7): 1309-1317. (YAN Jingsen, WANG Haiyan, ZHANG Jingru, et al. Effect of TiO2-Al2O3support preparation technique on hydrodenitrogenation of Ni2P/TiO2-Al2O3catalysts[J].Acta Physico-Chimica Sinica, 2014, 30(7): 1309-1317.)
[10] 柴永明, 安高军, 柳云骐, 等. 过渡金属硫化物催化剂催化加氢作用机理[J].化学进展, 2007, 19(2/3): 234-242. (CHAI Yongming, AN Gaojun, LIU Yunqi, et al. Transition metal sulfides hydrogenation catalysts: Active phase structure and mechanism of the catalytic reaction[J].Progress in Chemistry, 2007, 19(2/3): 234-242.)
[11] 高玉兰, 方向晨. 加氢处理催化剂器外预硫化技术研究与展望[J].化工进展, 2010, 29(3): 465-471. (GAO Yulan, FANG Xiangchen. Progress and prospect in ex-situ presulfurization for hydrotreating catalyst[J].Chemical Industry and Engineering Progress, 2010, 29(3): 465-471.)
[12] 罗树权, 孙征, 高雪. 加氢催化剂器外预硫化技术现状[J] . 化工技术与开发, 2014, 8(3): 34-37. (LUO Shuquan, SUN Zheng, GAO Xue. Current situation of ex-situ pre-sulfiding hydrotreatment catalyst[J].Technology & Development of Chemical Industry, 2014, 8(3): 34-47.)
[13] 丁庆玉, 于春梅, 王燕. 加氢催化剂器外预硫化技术研究[J].化学工程师, 2013, 27(7): 70-73. (DING Qingyu, YU Chunmei, WANG Yan. Research of off-site presulfurization hydrotreating catalysts[J].Chemical Engineer, 2013, 27(7): 70-73.)
[14] 李童, 董群, 冯熙桐. 加氢催化剂预硫化技术研究进展[J].化学工程师, 2014, 28(1): 42-44. (LI Tong, DONG Qun, FENG Xitong. Research progress in the hydrogenation catalyst presulfrization[J].Chemical Engineer, 2014, 28(1): 42-44.)
[15] GAO Y, FANG X, CHENG Z. A comparative study on the ex situ and in situ presulfurization of hydrotreating catalysts[J].Catalysis Today, 2010, 158: 496-503.
[16] 夏远亮. 原位分解法制备免预硫化CoMoS/γ-Al2O3催化剂的表征及加氢性能研究[J].分子催化, 2008, 22(3): 224-229. (XIA Yuanliang. Study on characterization and hydrogenization performance of CoMoS/γ-Al2O3, catalyst prepared by in-situ decomposition method[J].Journal of Molecular Catalysis (China), 2008, 22(3): 224-229.)
[17] 林凌, 伊晓东, 邱波, 等. 免预硫化加氢脱硫MoNiP/Al2O3催化剂的制备和表征[J].催化学报, 2007, 28(12): 1096-1100. (LIN Ling, YI Xiao-dong, QIU Bo, et al. Preparation and characterization of catalyst for tiophene presulfidation-free MoNiP/Al2O3hydrodesulfurization[J].Chinese Journal of Catalysis, 2007, 28(12): 1096-1100.)
[18] 田维乾, 刘静, 刘灿, 等. CoMoS/γ-Al2O3催化剂对麻疯树油加氢处理的研究[J].燃料化学学报, 2013, 41(2): 207-213. (TIAN Weiqian, LIU Jing, LIU Can, et al. Hydrotreatment of jatropha oil over CoMoS/γ-Al2O3catalyst[J].Journal of Fuel Chemistry and Technology, 2013, 41(2): 207-213.)
[19] 任春晓, 吴培, 李振昊, 等. 加氢催化剂预硫化技术现状[J].化工进展, 2013, 32(5): 1060-1064. (REN Chunxiao, WU Pei, LI Zhenhao, et al. The status of presulfurization technology for hydrogenation catalyst[J].Chemical Industry and Engineering Progress, 2013, 32(5): 1060-1064.)
[20] YU S H, WU Y S, YANG J, et al. A novel solventothermal synthetic route to nanocrystalline CdE (E=S, Se, Te) and morphological control[J].Chemistry of Materials, 1998, 10(10): 2309-2312.
[21] 吴雨航, 凌凤香, 赵国利, 等. CoMo/Al2O3-SiO2催化剂原位红外光谱研究[J].当代化工, 2015, 44(5): 962-965. (WU Yuhang, LING Fengxiang, ZHAO Guoli, et al. In-situ FT-IR Study on the CoMo/Al2O3catalyst[J].Contemporary Chemical Industry, 2015, 44(5): 962-965.)
[22] DELIY I V, VLASOVA E N, NUZHDIN A L, et al. Hydrodeoxygenation of methyl palmitate over sulfided Mo/Al2O3, CoMo/Al2O3and NiMo/Al2O3catalysts[J].Rsc Advances, 2013, 4(5): 2242-2250.
[23] 张玉涵, 凌凤香, 王少军, 等. Co-Mo/γ-Al2O3催化剂的原位红外光谱表征研究[J].燃料化学学报, 2013, 41(6): 710-714.(ZHANG Yuhan, LING Fengxiang, WANG Shaojun, et al. An in-situ FT-IR study on the CO and NO co-adsorption on the Co-Mo/γ-Al2O3catalysts[J].Journal of Fuel Chemistry and Technology, 2013, 41(6): 710-714.)
[24] HADJIVANOV K I, VAYSSILOV G N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule[J].Cheminform, 2003, 34(17): 307-511.
[25] 邱丽美, 齐和日玛, 刘清河, 等. X射线光电子能谱法研究加氢脱硫催化剂中活性元素的化学态[J].石油学报(石油加工), 2011, 27(4): 638-642. (QIU Limei, Qiherima, LIU Qinghe, et al. Investigation of chemical states for hydrodesulfurization catalysts by using X-Ray photoelectron spectroscopy[J].Acta Petrolei Sinica(Petroleum Processing Section), 2011, 27(4): 638-642.)
[26] 朱崇业, 牛国兴, 陈海鹰, 等. 钼镍系列加氢处理催化剂的表面活性结构[J].复旦学报(自然科学版), 1995, 34(5): 490-500.(ZHU Chongye, NIU Guoxing, CHEN Haiying, et al. Surface species structure of nickel-molybdenum hydrotreating catalyst[J].Journal of Fudan University (Natural Science), 1995, 34(5): 490-500.)
[27] 周同娜, 尹海亮, 柳云骐,等. 磷含量对NiMo/γ-Al2O3催化剂活性相结构的影响[J].燃料化学学报, 2010, 38(1): 69-74. (ZHOU Tongna, YIN Hailiang, LIU Yunji, et al. Effect of phosphorus on the interaction between active component and carrier of NiMo/Al2O3catalysts[J].Petroleum Processing and Petrochemicals, 2010, 38(1): 69-74.)
[28] FERDOUS D, DALAI A K, ADJAYE J, et al. Surface morphology of NiMo/Al2O3catalysts incorporated with boron and phosphorus: Experimental and simulation[J].Applied Catalysis A General, 2005, 294(1): 80-91.
[29] HUIRACHE-ACUA R, ALBITER M A, ESPINO J, et al. Synthesis of Ni-Mo-W sulphide catalysts by ex situ decomposition of trimetallic precursors[J].Applied Catalysis A General, 2006, 304(1): 124-130.
[30] FURIMSKY E, MASSOTH F E. Deactivation of hydroprocessing catalysts[J].Catalysis Today, 1999, 52(52): 381-495.
[31] LEBRETON R, BRUNET S, PÉROT G, et al. Deactivation and characterization of hydrotreating NiMo/Al2O3catalyst coked by anthracene[J].Studies in Surface Science & Catalysis, 1999, 126(99): 195-201.
Preparation, Characterization and Catalytic Hydrotreating Performance ofStepped Presulfurized Mo-Ni/γ-Al2O3Catalyst
ZHANG Qiang1, DING Wei1,3, WANG Dingcong2, ZHAO Dezhi1, YANG Chensi1, ZHANG Zhiwei1
(1.CollegeofChemicalEngineeringandEnvironmentalEngineering,LiaoningUniversityofPetroleum&ChemicalTechnology,Fushun113001,China; 2.FushunResearchInstituteofPetroleumandPetrochemicals,SINOPEC,Fushun113001,China;3.StateKeyLaboratoryofHeavyOilProcessing,ChinaUniversityofPetroleum,Beijing102249,China)
The prevulcanization precipitation liquid was prepared by self-assembly with thiocarbamide, citric acid and polyethylene glycol-200. The two-stepped presulfurization Mo-Ni/Al2O3catalysts were prepared by in-situ precipitation method and characterized by BET, XRD, FT-IR of CO adsorbed, XPS, and TEM. The hydrogenation performances of the prepared catalyst were evaluated with micro fixed bed reactor and with the directly in-situ presulfurization catalyst as reference. The results showed that the two-stepped presulfurization Mo-Ni/Al2O3already achieved 74.8% of sulfidity after the first step of presulfurization (ex-situ presulfurization). The additions of polyethylene glycol-200 and thiocarbamide made an improvement of the pore properties of the catalysts. The biggest specific surface of 198 cm2/g was obtained when the mass fraction of thiocarbamide reached 20% of Mo and Ni oxides in the catalyst, at the same time, the active metal dispersion was better and the sulfidity was 2.60 percentage points higher than the reference catalyst. The hydrodesulfurization (HDS), hydrodenitrogenation (HDN) and hydrodearomatization (HDA) rates of 40 h hydrotreating for FCC diesel fraction over the two-stepped presulfurization Mo-Ni/γ-Al2O3catalyst were 2.28 percentage points, 1.94 percentage points and 3.46 percentage points higher than those over the reference catalyst, respectively.
two-stepped presulfurization; in-situ precipitation; catalysts; hydrodesulfurization (HDS); hydrodenitrogenation (HDN)
2016-01-15
中国石油化工集团公司资助项目(总合-JQ1416)和中国海洋石油总公司资助项目(20140331)基金资助
张强,男,硕士研究生,主要从事加氢工艺及其催化剂研究
丁巍,女,讲师,博士研究生,从事重质油加氢催化剂研发;E-mail:cicy1125@163.com
1001-8719(2017)01-0032-10
TQ426.95
A
10.3969/j.issn.1001-8719.2017.01.005
