HPLC法测定阿法替尼片中主药和有关物质的含量
2017-01-16李赫宇
李赫宇,赵 玲
(1. 天津市益倍建生物技术有限公司,天津 300457; 2. 武汉轻工大学生物与制药工程学院,武汉 430023)
HPLC法测定阿法替尼片中主药和有关物质的含量
李赫宇1,赵 玲2*
(1. 天津市益倍建生物技术有限公司,天津 300457; 2. 武汉轻工大学生物与制药工程学院,武汉 430023)
目的:建立以高效液相色谱法测定阿法替尼片中主药和有关物质含量的方法,以控制该制剂的质量。方法:色谱柱为Kromasil C18(250 mm×4.6 mm,5 μm),流动相为乙酸铵水溶液(30 mmol/L)-甲醇(25∶75),流速为1.0 ml/min,紫外检测波长为254 nm,柱温30 ℃,进样量为20 μl。结果:阿法替尼检测浓度的线性范围为60.13~600.45 μg/ml(r=0.999 6);平均回收率为99.41%,RSD为0.86%。结论:建立的检测方法简单,结果准确可靠,可用于阿法替尼片的质量控制。
阿法替尼,含量测定,高效液相色谱法,有关物质
阿法替尼是由德国勃林格殷格翰公司研发的多靶点抗癌新药,于2013年通过美国食品药品监督管理局(FDA)的审批,用以治疗非小细胞肺癌、大肠癌、乳腺癌和头颈癌的分子靶向制剂,商品名为Tovok/Gilotrif[1]。肺癌是临床上最常见的恶性肿瘤,且肺癌患者里约85%为非小细胞肺癌,严重危害人类的生命健康,因此该药一经问世很快成为晚期非小细胞肺癌的一线治疗新药[2]。阿法替尼属于多靶点酪氨酸激酶(TK) 抑制剂,是TK的不可逆性抑制剂,也是人类表皮生长因子受体(EGFR)抑制剂[包括人类表皮生长因子受体2(HER2)、HER4两个亚族]。阿法替尼片剂的主药以阿法替尼马来酸盐的形式存在。《美国药典》中尚未收载阿法替尼的含量测定方法,为此,本文尝试建立高效液相色谱(HPLC)法测定阿法替尼马来酸盐和有关物质含量测定的方法,为阿法替尼片的质量控制,以及评价其质量的一致性提供可参考的依据。
1 仪器与试药
美国agilent1260高效液相色谱系统;甲醇(美国Tedia公司产品,色谱纯);乙酸铵(天津市天力化学试剂有限公司产品,分析纯)。阿法替尼片(武汉轻工大学制剂室,批号150801、150802、150803,规格:每片含阿法替尼马来酸盐59.12 mg,相当于每片含阿法替尼40 mg);阿法替尼对照品(武汉轻工大学生物与制药工程学院,批号150922,质量分数:99.82%); 阿法替尼合成方法参考文献完成[3]。
2 方法与结果
因阿法替尼马来酸盐在含水溶液中解离成马来酸和阿法替尼,马来酸为不饱和烯酸,极性较强,在ODS柱上保留时间短,先出峰,阿法替尼为有机碱,极性相对较弱,后出峰。本实验按外标法以阿法替尼来计算含量。
2.1 色谱条件 色谱柱:Kromasil C18(250 mm×4.6 mm,5 μm),流动相为乙酸铵水溶液(30 mmol/L)-甲醇(25∶75),体积流量1.0 ml/min,紫外检测波长为254 nm,柱温25 ℃,进样量20 μl。
2.2 溶液制备
2.2.1 对照品溶液 取阿法替尼对照品,用流动相溶解,并制成阿法替尼质量浓度为505.75 μg/ml的溶液,作为对照品溶液。
2.2.2 供试品溶液 取阿法替尼片剂研细为粉末适量,约相当于阿法替尼12.643 75 mg,精密称定,放置于25 ml量瓶中,用流动相乙酸铵水溶液-甲醇溶解并稀释至刻度,滤过,取续滤液作为供试品溶液。
2.2.3 有关物质溶液制备 取阿法替尼片粉末适量,用流动相配制成质量浓度为20 μg/ml的溶液作为供试液。再用流动相稀释,配制成质量浓度为0.2 μg/ml的对照液。
2.3 系统适应性试验 精密量取“2.2.1”项下对照品溶液、供试品溶液各20 μl,按照“2.1”项下色谱条件注入液相色谱仪,记录峰面积,并以流动相溶液乙酸铵水溶液(30 mmol/L)-甲醇(25∶75)作为参照。以阿法替尼峰面积计算,理论塔板数为8 478,保留时间为11.784 min,峰形对称,见图1。
2.4 线性关系与检测限考查 精密称取阿法替尼对照品适量,用流动相溶解并稀释成阿法替尼质量浓度分别为60.13、149.89、300.19、451.09和600.45 μg/ml的系列溶液,精密量取各溶液20 μl分别注入液相色谱仪,记录色谱图。以进样的质量浓度(C)为横坐标,峰面积(A)为纵坐标进行线性回归,得回归方程A=1.018 5C+4.1315(r=0.999 6)。结果表明,阿法替尼质量浓度与峰面积的线性关系良好,线性范围为60.13~600.45 μg/ml。以信噪比S/N=3计算,阿法替尼检测限为0.4 ng。
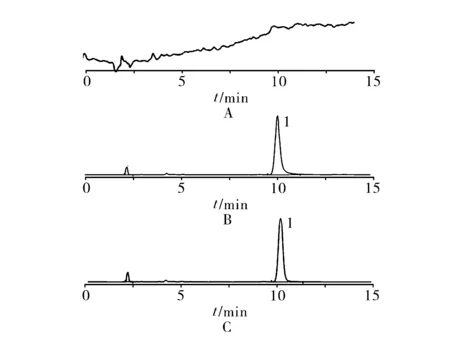
1.阿法替尼
2.5 精密度试验 取阿法替尼对照品适量,按照“2.2.1”项下方法制备对照品溶液,精密量取20 μl按照“2.1”项下色谱条件,连续进样5次进行测定。结果显示峰面积积分值的RSD为0.68%,表明本方法精密度良好。
2.6 重现性试验 精密称取同一批号(批号为150802)的阿法替尼样品6份,分别按照“2.2”项下的方法制备供试品溶液和对照品溶液,分别量取20 μl注入液相色谱仪,依“2.1”项下色谱条件下进行测定。结果显示阿法替尼的平均质量分数为99.18%,RSD为0.24%,表明本方法重现性良好。
2.7 稳定性试验 取制备的供试品溶液1份,分别在0、1、2、4和8 h量取20 μl注入液相色谱仪,依“2.1”项下色谱条件测定,峰面积积分值的RSD为0.58%,结果表明供试品溶液在8 h内稳定。
2.8 回收率试验 取同一批号的样品(批号150801),按测试浓度的80%、100%和120% 3个梯度制备供试品溶液,分别加入一定质量的阿法替尼对照品,每个浓度各取20 μl的3份样品注入液相色谱仪,按照 “2.1”项下色谱条件进样测定,记录峰面积,并计算回收率。结果平均回收率为99.41%,RSD为0.86%,见表1。
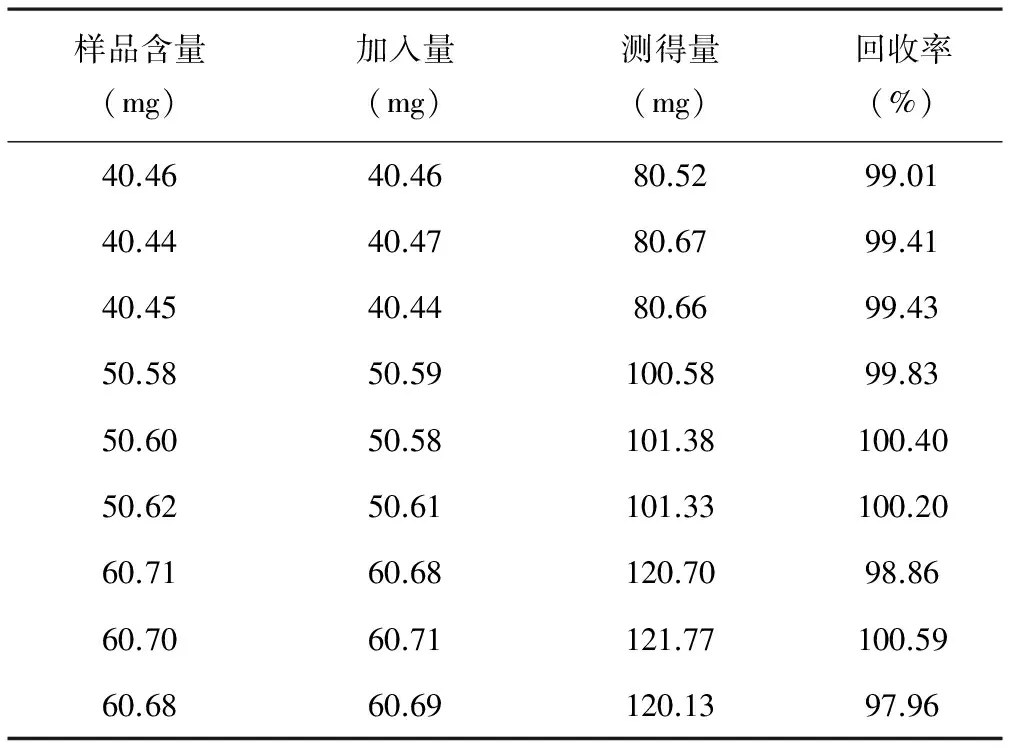
表1 回收率试验结果(n=9)
2.9 中间体分离试验及破坏性试验 取阿法替尼对照品及中间体6-氨基-4-[(3-氯-4-氟-苯基)氨基]-7-[(S)-(四氢呋喃-3-基)氧基]喹唑啉(中间体Ⅰ)适量,用流动相配制成阿法替尼质量浓度为20 μg/ml,中间体Ⅰ质量浓度为2 μg/ml的溶液,作为中间体分离试验的供试溶液。另取阿法替尼片剂粉末(约相当于阿法替尼200 μg)5份,分别放置于10 ml的量瓶中。第1份量瓶里加入1 ml物质的量为0.1 mol/L的盐酸溶液;第2份量瓶里加入1 ml物质的量为0.1 mol/L的氢氧化钠溶液;第3份量瓶里加入1 ml 3%的H2O2溶液。静置2 h后,第1份量瓶里加入0.1 mol/L的氢氧化钠溶液1 ml酸碱中和后,加流动相稀释至刻度,滤过,取续滤液作为酸破坏的供试品溶液;第2份量瓶里加入0.1 mol/L的盐酸1 ml酸碱中和后,加流动相稀释至刻度,滤过,取续滤液作为碱破坏的供试品溶液;第3份量瓶里加流动相稀释至刻度,滤过,取续滤液作为氧化破坏的供试品溶液;第4份量瓶置于4 000 lx的光下照射6 h;第5份量瓶置于60 ℃水浴中加热2 h。分别量取上述各量瓶里的供试品溶液20 μl注入液相色谱仪,按照“2.1”项下色谱条件进行检测,记录色谱图。结果中间体化合物Ⅰ的峰与阿法替尼峰能完全分离;5种极端条件下(酸、碱、氧化、光照和高温破坏)均有降解产物产生,而且5种降解产物都能与阿法替尼峰完全完分离。中间体分离试验与5种极端条件破坏后的色谱见图2。
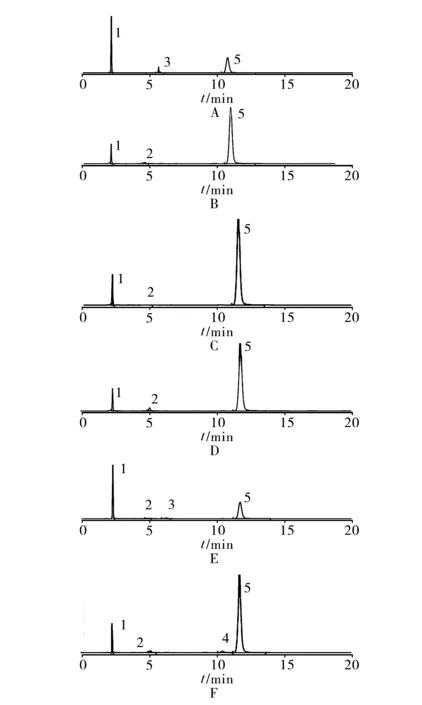
1.马来酸 2.杂质Ⅰ 3.中间体Ⅰ 4.杂质Ⅱ 5.阿法替尼
2.10 样品含量测定 取阿法替尼片粉末、阿法替尼对照品适量,按“2.2”项下方法分别制备供试品溶液和对照品溶液。分别精密量取20 μl注入液相色谱仪,按照 “2.1”项下色谱条件进样测定,记录峰面积,并按外标法以阿法替尼和阿法替尼马来酸盐的峰面积,计算含量,结果见表2。

表2 样品含量测定结果
2.11 样品有关物质测定 取阿法替尼片粉末适量,按“2.2”项下方法制备成供试液和对照液,量取对照液20 μl进样,调节仪器检测灵敏度,使阿法替尼峰峰高为满量程的10%~30%;再量取供试液20 μl进样,记录色谱至主峰保留时间的3 倍,记录主峰和有关物质的峰面积,按1%自身对照法计算有关物质的含量。结果3批样品中有关物质含量分别为0.19%、0.21%和0.20%。
3 讨论
阿法替尼是FDA继吉非替尼、厄洛替尼和拉帕替尼批准上市的第4个分子靶向EGFR抑制剂,其是可逆性ATP竞争性抑制剂,已经被确认为治疗具有19和21位EGFR突变的晚期非小细胞肺癌最有效的手段,可提高肿瘤患者的生存期的生活质量。但是,对其主药的含量和有关物质测定方法还鲜有报道。探讨其主要成分的质量控制方法,可以提高进口药与国产药的质量一致性,为提高制剂水平提供依据。
3.1 检测波长的选择 前期进行了预实验,分别称取阿法替尼对照品和阿法替尼片剂粉末适量,用流动相乙酸铵水溶液-甲醇配制成质量浓度约505.75 μg/ml的对照品溶液、供试品溶液,按照最新版《中国药典》规定的紫外分光-光度法[5]操作,于200~400 nm波长内进行检测。结果显示对照品溶液、供试品溶液均于254 nm处有最大吸收,且辅料溶液在此处无吸收,故检测波长选为254 nm。
3.2 流动相的选择 通过反复配制不同浓度、不同pH的乙酸铵溶液,同时不断改变甲醇与乙酸铵溶液的比例,经过多次调整,最后发现当乙酸铵水溶液浓度为30 mmol/L,与甲醇的比例为25∶75时,阿法替尼的主峰保留时间合适,峰形比较对称,其与中间体、有关物质和降解产物均能得到比较好的分离,该方法具有较强的专属性,良好的准确度和精密度,溶液在8 h内也稳定。因此,最终确定使用75∶25的甲醇-30 mmol/L乙酸铵水溶液作为流动相。
1 刘丹,栾天,袁莹,等. 多靶点酪氨酸激酶抑制剂阿法替尼及其类似物的研究进展[J]. 中国药学杂志,2014,49(24):2145-2149
2 安富荣,王淑萍,归小龙. 晚期非小细胞肺癌一线治疗新药阿法替尼[J] .中国药师,2015,18(1):136-138
3 李晴晴,张庆文. 阿法替尼的合成路线图解 [J] .中国医药工业杂志, 2015,46(4):422-425
4 Singer T, Colbatzky F, Platz S. Utilization of inhibitors of EGFR-mediated signal transduction for the treatment of benign prostatic hyperplasia (bph)/prostatic hypertrophy: WO, 2003094921A2 [P]. 2003-11-20
5 中国药典[S].二部.2010:附录38
药物与临床
Quantitative determination of main component and related substances in afatinib tablets by HPLC
Li Heyu1, Zhao Ling2
(1.Tianjin Ubasic Health Nutrition Co Ltd,Tianjin 300457;2. College of Pharmaceutical Engineering,Wuhan Polytechnic University,Wuhan 430023)
Objective: To establish an HPLC method for the determination of the main component and related substances in afatinib tablets.Methods: The determination was performed on Kormasil C18column (250 mm×4.6 mm, 5 μm) with mobile phase composed of ammonium acetate solution(30 mmol/L)-methanol(25∶75) at a flow rate of 1.0 ml/min. The detection wavelength was set at 254 nm at 30 ℃. The injection size was at 20 μl.Results: The calibration curve of afatinib was linear in the range of 60.13~600.45 μg/ml(n=5,r=0.999 6),and the average recovery of afatinib was 99.41%(RSD was 0.86%).Conclusions: This method is rapid,accurate and reliable,and applicable for the quality control of afatinib tablets.
afatinib,content determination,HPLC,related substances
2016-05-27
*通讯作者:赵玲,E-mail:zhaolingcpu@126.com。
R927.2
A
1006-5687(2016)04-0022-04
