迟缓爱德华氏菌Eta1-L-Gapdh融合蛋白在蓝藻中的表达
2017-01-03张泽峰王晶晶王春梅
王 智, 张泽峰, 王晶晶, 王春梅
(北京中医药大学 生物制药系,北京 100102)
迟缓爱德华氏菌Eta1-L-Gapdh融合蛋白在蓝藻中的表达
王 智, 张泽峰, 王晶晶, 王春梅*
(北京中医药大学 生物制药系,北京 100102)
在蓝藻中表达迟缓爱德华氏菌Eta1-L-Gapdh融合蛋白。提取迟缓爱德华氏菌基因组DNA为模板,用PCR技术分别扩增两个已知具有较强免疫原性的基因eta1和gapdh,再采用重叠延伸PCR将这两个基因融合,获得目的融合基因eta1-L-gapdh。将目的基因连接到表达载体pRL489的两个BamH I酶切位点之间构建表达载体,用质粒提取、PCR、酶切、测序等手段对表达载体进行验证。验证正确的表达载体通过三亲接合转化野生鱼腥藻PCC7120,用新霉素抗性筛选出转基因藻落,通过质粒提取和PCR验证转基因藻。用RT-PCR和Western-blot分别从转录水平和翻译水平对转基因藻中融合基因的表达进行了检测。结果表明,含目的基因的表达载体构建成功,目的基因在蓝藻中转录并表达蛋白,该蛋白在蓝藻中的表达量为2.46%。
迟缓爱德华氏菌;eta1;gapdh;鱼腥藻PCC7120;三亲接合;融合蛋白
迟缓爱德华氏菌(Edwardsiellatarda)是一种危害性极大的致病菌[1],可以感染包括淡水与海水养殖的多种鱼类,并能在短期内引起鱼类大量死亡,近年来给我国水产养殖业造成严重损失[2]。科学家希望尽快研制疫苗以预防控制该疾病[3]。目前针对该菌的疫苗研究已有大量的文献报道,主要来自于国内[4-9],且研究的重点方向之一是寻找具有较强免疫原性的蛋白。自从2004年日本学者Kawai等[10]发现37 kDa的迟缓爱德华氏菌外膜蛋白具有较强的免疫原性后,越来越多的蛋白被证明能对迟缓爱德华氏菌起到免疫保护作用。到目前为止,至少已经发现17种不同的蛋白具有较强的免疫原性,具有开发成疫苗的潜力。注射、浸泡和口服是鱼类疫苗的主要接种方式[11]。其中口服方式实施简单,但在发挥效应前需要防止消化道酶的降解和肠上皮细胞胞饮作用的降解[12]。因此寻找合适的载体保护疫苗蛋白是开发口服疫苗需要解决的关键问题之一。蓝藻是一种能进行光合产氧的原核生物[13],因其具有细胞壁,因此可以考虑将其用作疫苗的载体避免口服疫苗被鱼类胃肠道消化酶降解;生产疫苗的蓝藻还可作为鱼类的理想食物直接喂养鱼类,这也表明其适合开发成口服疫苗;同时其结构简单,生长迅速,适应性强,易于进行遗传操作,用于生产疫苗的成本较低;且蓝藻多数不含毒蛋白,用于生产疫苗非常安全[14]。邓元告、Jia等[15-16]已经利用蓝藻表达对虾白斑病毒VP28基因,将转基因藻喂食对虾后取得了较好的免疫保护作用。但目前国内外未见利用蓝藻表达鱼类病害基因用以开发口服疫苗的报道。本研究采用鱼腥藻PCC7120(Anabaenasp. PCC7120)表达迟缓爱德华氏菌的两个具有较强免疫原性基因的融合基因,并从转录和翻译水平检测融合蛋白的表达,以期为该病的疫苗研发奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌种、藻种与质粒 迟缓爱德华氏菌HtD来源于人工养殖的大菱鲆(ScophthalmusmaximusL.),由山东烟台莱州大华水产有限公司杨学宋经理惠赠;野生型鱼腥藻PCC7120由中国科学院过程工程研究所丛威教授惠赠;大肠埃希菌HB101由本实验室保存;大肠埃希菌ED8654由美国弗吉尼亚联邦大学Jeff Elhai教授惠赠;三亲接合所用辅助质粒pRL623由美国密歇根州立大学Peter Wolk教授惠赠;克隆载体pEasy Blunt Zero及感受态细胞Trans T1购自北京全式金生物技术有限公司。
1.1.2 培养基 LB培养基(g/L):胰蛋白胨10,酵母浸膏5,NaCl 10,根据需要添加100 μg/mL氨苄青霉素或50 μg/mL卡那霉素;BG-11培养基配方参照文献[17]。
1.1.3 主要试剂 Pfu DNA聚合酶、Anti-His单克隆抗体、核酸及蛋白分子Marker购自北京全式金生物技术有限公司;BamH I、T4 DNA 连接酶、EcoR I、热敏磷酸酶均购自New ENGLAND Biolabs (NEB)公司;6×DNA Loading Buffer、DNAase I、cDNA第1链合成试剂盒购自TaKaRa公司;2×TaqPCR Master Mix、胶回收试剂盒、质粒提取试剂盒均购自北京博迈德基因技术有限公司;底物发光试剂盒、二抗Anti-Ms Ig(H+L)/HRP购自北京鼎国昌盛生物技术有限公司;Trizol试剂购自Invitrogen公司;实验所用其他生化试剂购自北京拜尔迪生物技术有限公司,均为分析纯;PCR引物合成及序列测定均由北京博迈德基因技术有限公司完成。
1.2 方法
1.2.1 迟缓爱德华氏菌基因组DNA提取 实验方案参照精编分子生物学实验指南(第4版)进行[18]。实验结束后取5 μL基因组DNA与1 μL 6×DNA Loading Buffer混合,进行1%的琼脂糖凝胶电泳,检测提取结果。
1.2.2 PCR扩增免疫原性蛋白基因eta1及gapdh在NCBI上检索获得迟缓爱德华氏菌免疫原性蛋白基因eta1及gapdh的基因序列,其登录号分别为JN802138.1和FJ605131.1,根据这两个序列设计用于扩增这两个基因的上下游引物,为了用pRL 489载体表达,在eta1上游引物的5′端添加一段SD序列GGAGAG,同时根据需要,在引物的5′端添加合适的酶切位点。扩增eta1基因用eta1F-eta1R引物对,扩增gapdh基因用gapdhF-gapdhR引物对,引物信息见表1。用全式金公司的TransStart Fast Pfu DNA 聚合酶进行扩增,eta1基因反应条件为95 ℃ 2 min,(95 ℃ 20 s,55 ℃ 20 s,72 ℃ 15 s,35个循环),72 ℃ 5 min;gapdh基因的反应条件为95 ℃ 2 min,(95 ℃ 20 s,52 ℃ 20 s,72 ℃ 40 s,35个循环),72 ℃ 5 min。反应结束后用2%的琼脂糖凝胶电泳检测PCR结果。
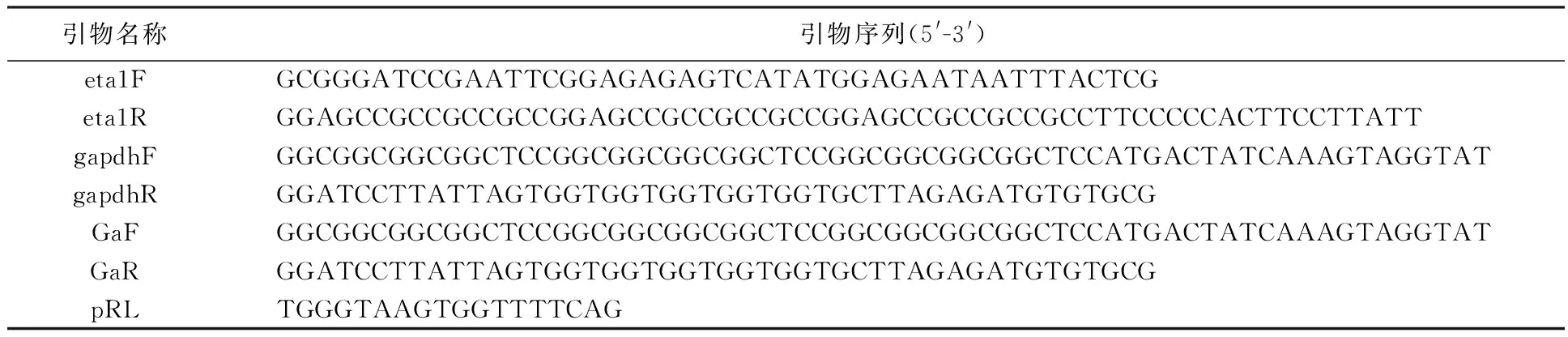
表1 实验所用部分引物信息Table 1 Primers used in this study
1.2.3 重叠PCR扩增融合基因(eta1-L-gapdh) 将gapdh和eta1的PCR产物胶回收,分别取1 μL胶回收后的产物作模板,进行重叠延伸反应,选择80 ℃作为退火温度,不加入引物,但加入相应浓度的dNTPs、Mg2+和Pfu DNA聚合酶并补水至20 μL,72 ℃延伸70 s,共进行10个循环;第一阶段的延伸结束后,从体系中取出1 μL作模板,用Pfu DNA 聚合酶进行普通PCR反应。普通PCR反应条件为95 ℃ 2 min,(95 ℃ 20 s,55 ℃ 20 s,72 ℃ 70 s,35个循环),72 ℃ 5 min。反应结束后用2%的琼脂糖凝胶电泳检测PCR结果。
1.2.4 重组表达载体构建 将所得融合基因胶回收纯化后,按pEASY-Blunt Zero克隆载体说明书进行转化实验,培养阳性转化子,用试剂盒提取质粒进行PCR验证并用M13通用测序引物测序验证。验证正确后用BamH I酶切含有目的基因的克隆质粒,回收目的片段。同时,培养含pRL489载体的HB101菌株,提取质粒用BamH I将其酶切,回收大片段用于连接。将酶切后胶回收得到的pRL489载体大片段用热敏磷酸酶按说明书进行去磷酸化,反应条件为37 ℃水浴30 min,再65 ℃水浴5 min热失活;再用酚氯仿抽提纯化。用T4 DNA连接酶将pRL489大片段与融合基因片段进行连接,连接反应用PCR仪控温,在25 ℃下连接1.5 h;取连接后产物10 μL于大肠埃希菌HB101感受态细胞中,进行常规转化实验。转化后的阳性克隆用PCR、BamH I单酶切、EcoRI单酶切、测序等手段验证。
1.2.5 重组表达载体转化鱼腥藻PCC7120 将培养于固体BG-11培养基上的野生鱼腥藻PCC7120接种到液体培养基中摇床震荡培养,转速140 r/min,培养温度28 ℃,同时24 h提供光照强度为3 000~4 000 lx的白光。待其生长至对数期,参照文献[19]的方法,将其和含有表达载体的大肠埃希菌HB101以及含有辅助质粒和接合质粒的大肠埃希菌ED8654混合,将混合液涂于贴在含BG-11固体培养基平皿上的滤膜表面。将平皿置于培养箱中培养。
1.2.6 转基因鱼腥藻的筛选和验证 上述平皿培养2 d后,将滤膜转移至含40 μg/mL的新霉素的固体BG-11培养基平皿上正常培养至藻落可见。挑取藻落于25 mL含30 μg/mL新霉素的BG-11液体培养基中继续筛选。参照文献[20]的方法提取转基因藻的质粒,以所提质粒为模板,PCR验证pRL489载体与eta1基因相连部分序列、eta1基因、gapdh基因及融合基因。所用引物对分别为pRL-eta1R引物对、eta1F-eta1R引物对、GaF-GaR引物对和eta1F-GaR引物对。引物信息见表1。PCR反应程序分别为94 ℃ 8 min,(94 ℃ 30 s,50 ℃ 30 s,72 ℃ 65 s,30个循环),72 ℃ 5 min;94 ℃ 8 min,(94 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,30个循环),72 ℃ 5 min;94 ℃ 8 min,(94 ℃ 30 s,55 ℃ 30 s,72 ℃ 65 s,30个循环),72 ℃ 5 min;94 ℃ 8 min,(94 ℃ 30 s,55 ℃ 30 s,72 ℃ 110 s,30个循环),72 ℃ 5 min;反应结束后进行电泳。
1.2.7 RT-PCR检测融合基因表达 培养转基因藻至对数期,用Trizol试剂提取转基因藻和野生藻的总RNA,操作按照Trizol试剂说明书进行。RNA提取后向体系中加入DNAase I,37 ℃水浴反应30 min以除去其中痕量的DNA,再用pH<5.0的酚氯仿将其纯化。按照PrimeScript 1st Strand cDNA Synthesis kit说明书进行反转录反应,反转录结束后用2×TaqPCR Master Mix进行普通PCR反应。用rnpBF-rnpBR引物对扩增蓝藻内参基因rnpB以验证所提RNA的完整性,同时用eta1F-eta1R引物对(见表1)、EF-ER引物对、GF-GR引物对、EGF-EGR引物对分别检测完整eta1基因、部分eta1基因、部分gapdh基因以及eta1基因与gapdh基因的连接序列(简称eg)的转录情况。所用引物信息见表2。PCR反应程序均采用94 ℃ 8 min,(94 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,30个循环),72 ℃ 5 min;反应结束后进行电泳。

表2 RT-PCR所用引物情况Table 2 Primers used for RT-PCR
1.2.8 蓝藻总蛋白提取 取培养至对数后期颜色深绿的转基因藻和野生藻10 mL于15 mL离心管中,2 000 r/min离心2 min沉淀藻细胞;小心弃去上清后将蓬松的藻细胞沉淀转移至1.5 mL离心管中;10 000 r/min离心1 min,弃上清,加入100 μL蒸馏水,稍混匀后进行超声反复冻融,流程为-80 ℃冻10 min,室温超声3 min,重复5次;12 000 r/min离心5 min收集上清即为蓝藻总蛋白。
1.2.9 Western-blot检测融合蛋白表达 取蓝藻总蛋白15 μL与3 μL 6×蛋白Loading Buffer混合煮沸5 min,冰浴2 min,10 000 r/min离心1 min吸取上清上样进行SDS-PAGE电泳,电泳条件设置为80 V/150 V,即开始电压为80 V,待样品浓缩到分离胶与浓缩胶界限处时将电压调整至150 V,待染料泳动到距离玻璃板下边缘1 cm处停止电泳;电泳结束后进行常规Western-blot检测,一抗为Anti-His单克隆抗体,二抗为辣根过氧化物酶标记抗小鼠多克隆抗体,稀释比例均为1∶5 000。
1.2.10 融合蛋白表达量的确定 利用蛋白浓度为0.14 mg/mL的含组氨酸标签蛋白与蛋白浓度为6.9 mg/mL的含融合蛋白的蓝藻总蛋白样品共同上样进行Western-blot实验,实验结束后,扫描实验结果,用Quantity One 4.62软件对结果进行分析,比对其灰度值,根据灰度值的比值应与蛋白量的比值相同,确定藻总蛋白中融合蛋白的浓度;计算融合蛋白表达量:融合蛋白表达量=藻液中融合蛋白浓度/藻总蛋白浓度。实验重复3次,结果取平均值。
2 结果与分析
2.1 目的基因的获得
迟缓爱德华氏菌基因组DNA提取结果见图1a。由图1a可知,样品条带大于10 kb,条带单一明亮,无杂带及弥散条带,提取效果较好。利用所提取的基因组DNA做模板,先用普通PCR分别扩增获得迟缓爱德华氏菌eta1基因和gapdh基因,结果见图1b。图1b中eta1与gapdh所属的泳道条带单一明亮,eta1条带介于700 bp和800 bp之间,与其理论大小(750 bp)一致;gapdh大小约为1 000 bp,与其理论大小 (1 068 bp ) 一致 ;再利用事先设计好的连接肽序列(GGGGS)3作为桥梁,采用重叠PCR扩增目的融合基因,结果如图1c所示。图1c中条带明亮单一,大小与预期相符,表明融合基因eta1-L-gapdh扩增成功。

图1 目的融合基因扩增结果Fig.1 PCR amplification of the target fusion geneM1:1 kb DNA Marker1;M2:DNA Marker2;HtD:迟缓爱德华氏菌基因组DNA;gapdh:gapdh基因扩增结果;eta1:eta1基因扩增结果;rh:融合目的基因扩增结果M1: 1 kb DNA Marker1;M2: DNA Marker2; HtD: genome DNA of Edwardsiellatarda; gapdh: PCR amplification of gapdh gene; eta1: PCR amplification of eta1 gene; rh: PCR amplification of the target fusion gene
2.2 重组表达载体构建
构建成功的表达载体经过质粒提取、PCR验证目的基因、酶切验证目的基因克隆方向等多层次的验证,结果如图2所示。图2a为质粒提取结果;图2b为目的融合基因PCR验证结果,其中条带明亮单一,大小与预期相符;图2c为BamH I和EcoR I单酶切结果,BamH I单酶切显示两条带,大小与预期一致,EcoR I单酶切结果显示除一条大于10 kb的条带还有3条较小条带,结合pRL489载体本身特点及目的融合基因序列,可知条带均与预期相符,且同时表明目的基因连接的方向与预期一致。经过上述初步验证后,设计测序引物,将表达载体送往公司测序,将所测得序列与理论序列进行Blast比对,发现两者完全一致。由此表明含目的融合基因的表达载体构建成功。

图2 表达载体验证结果Fig.2 Verification of the expression vectorM1:DNA Marker 1;M2:DNA Marker2;P:表达载体质粒;rh:融合基因PCR验证结果;E:EcoR I单酶切表达载体结果;B:BamH I单酶切表达载体结果M1: DNA Marker1; M2: DNA Marker2; P: the expression vector; rh: Verification of the fusiongene; E: the result of EcoR I digestion of the expression vector;B: the result of BamH I digestion of the expression vector
2.3 转基因鱼腥藻的获得
将构建好的表达载体通过三亲接合转化野生鱼腥藻PCC7120后,用新霉素在固体平板和液体培养基中筛选阳性藻落(图3a和3b)。经过抗性筛选出的藻落用PCR进一步验证目的融合基因和表达载体,结果见图3c。图中各条带单一明亮,大小与预期一致,表明表达载体已成功转入野生鱼腥藻中。

图3 转基因鱼腥藻(a和b)及PCR验证结果(c)Fig.3 The transgenic cyanobacterium (a and b) and the results of PCR verification (c)A:三亲接合对照:野生鱼腥藻PCC7120;B:三亲接合后转基因鱼腥藻;M1:DNA Marker1;M2:DNA Marker2;1~4分别为:融合基因、pRL489载体与eta1基因相连部分序列、eta1基因、gapdh基因PCR验证结果A: wide-type Anabaena sp. PCC7120 as control; B: transgenic cyanobacteria;M1: DNA Marker1; M2: DNA Marker2; 1-4: fusion gene, the conjoint part of pRL489 and eta1 gene, eta1 gene,gapdh gene
2.4 目的融合基因的转录水平检测
从转录水平对目的融合基因的表达情况进行的检测结果见图4a和4b。图4a为蓝藻中内参基因rnpB RT-PCR扩增结果,产物条带单一明亮,大小正确,表明所提取的蓝藻总RNA完整性较好,图4b为各基因RT-PCR扩增结果,包括完整的eta1基因、部分eta1基因、部分gapdh基因、eta1基因与gapdh基因连接处序列,其大小均和理论相符,同时泳道5中野生型鱼腥藻中eta1基因片段RT-PCR无结果,泳道6中直接以所提总RNA进行PCR反应扩增eta1基因片段无结果,表明总RNA中无DNA残留,上述事实表明目的融合基因在鱼腥藻中确实进行了转录。
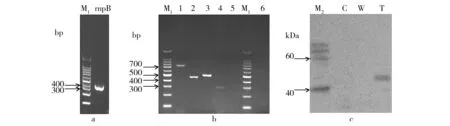
图4 融合基因转录(a和b)及翻译水平(c)检测结果Fig.4 Detection of expression of the target fusion gene at transcription (a and b) and translation level (c)M1:DNA Marker;M2:蛋白质Marker;rnpB:蓝藻内参基因;1~4:分别为以转基因藻总RNA为模板,eta1完整基因、eta1基因片段、eta1基因与gapdh基因连接部分序列、gapdh基因片段RT-PCR结果;5:以野生藻总RNA为模板,eta1基因片段RT-PCR结果;6:以转基因藻总RNA为模板,eta1基因片段PCR结果; C、W、T:分别为含有pRL489空载体转基因藻、野生藻、含目的基因转基因藻总蛋白Western-blot检测结果M1: DNA Marker; M2: protein Marker; rnpB: cyanobacteria reference gene; 1-4: RT-PCR results of intact eta1 gene, partial eta1 gene, the conjoint part of eta1 gene and gapdh gene, partial gapdh gene, respectively, using total RNA of transgenic cyanobacteria as the template; 5: RT-PCR result ofpartial eta1 gene by using total RNA of wide-type cyanobacteria as the template; 6: RT-PCR result of partial eta1 gene by using total RNA of transgenic cyanobacteria as the template; C: Western-blot detection of transgenic cyanobacteria bearing pRL489 vector; W: Western-blot detection of wide-type cyanobacteria bearing pRL489;T:Western-blot detection of transgenic cyanobacteria bearing the target fusion gene
2.5 目的融合基因的蛋白表达检测
目的融合基因在翻译水平的检测结果见图4c,与含有pRL489空载体转基因藻及野生藻泳道相比,图中含有目的基因的转基因藻泳道出现一条特异条带,表明融合蛋白得到了表达。经过比对灰度值后取平均值,得到融合蛋白浓度与标准蛋白浓度的比值为1.21。由此得到融合蛋白浓度为0.17 mg/mL。再根据1.2.10中公式得到目的蛋白在蓝藻中的表达量约为2.46%。
3 讨 论
本研究希望用蓝藻表达具有免疫原性的迟缓爱德华氏菌蛋白,为日后口服疫苗的开发奠定基础,因此需要合理选择免疫原性蛋白。本研究选用的两个蛋白分别来自迟缓爱德华氏菌的GAPDH和ETA1蛋白。其中GAPDH大小为37 kDa左右,是迟缓爱德华氏菌的一种外膜蛋白,具有较好的免疫保护作用[21]。该蛋白是最早从迟缓爱德华氏菌中发现的具有免疫原性的蛋白[10],之后又有多个研究小组对其保护作用进行了验证[22-25],在此期间还发现该蛋白对其他病菌也能起到较好的免疫作用[23-25]。本研究选择该蛋白作为抗原蛋白之一可以保证疫苗的免疫原性,同时也可以增加疫苗对其他病菌的适用性。ETA1理论分子大小为23.6 kDa,是一种细菌黏附素,也属于外膜蛋白,该蛋白用作亚单位疫苗时,对日本牙鲆(Japaneseflounder)的保护率高达83.3%[26]。同时,本研究首次将这两个蛋白用一段柔性连接肽连接,表达融合蛋白,希望可以进一步增强疫苗的免疫原性和保护作用。具体到疫苗的实施,首先需要大规模培养转基因藻,离心收集藻体并低温冻干,按照合适的剂量投入水体直接饲喂健康幼鱼,或者根据需要,可以向藻体中添加适当的疫苗佐剂,并通过多次间断饲喂加强疫苗的免疫保护作用。
本研究采用蓝藻中常用的表达载体pRL489[27]表达目的蛋白,pRL489载体上本身在两个BamH I位点之间含有luxAB报告基因。本研究将该报告基因替换成融合目的基因,同时在目的基因上游端添加了优化的SD序列[28]以增加表达量。根据实验设计本研究表达的融合蛋白理论分子量应为60.5 kDa,但实际检测到的分子量比该值要小,接近50 kDa。造成这一现象可能有以下原因:①融合蛋白被宿主体内的酶剪切断,剩下一段携带有组氨酸标签,因此可以通过Western-blot检测到;②由于改变了SD序列,引起了核糖体与mRNA结合位点的改变,翻译起始密码子的位置也由此改变,导致最终表达产物分子量的改变。分析eta1基因序列,发现除了N端起始密码子外,其序列内部另有2个甲硫氨酸密码子,若分别以这两个密码子作为起始密码子,表达的融合蛋白理论分子量大小分别是44和51.5 kDa,这样其中的51.5 kDa可能和实验中检测到的融合蛋白分子量大小相近。
[1] Mohanty B R,Sahoo P K.Edwardsiellosis in fish: a brief review[J].Journal of biosciences, 2007, 32(7): 1331-1344.
[2] Xiao J, Wang Q, Liu Q, et al. Isolation and identification of fish pathogenEdwardsiellatardafrom mariculture in China[J]. Aquaculture research, 2008, 40(1): 13-17.
[3] Choi S H,Kim K H.Generation of two auxotrophic genes knock-outEdwardsiellatardaand assessment of its potential as a combined vaccine in olive flounder (Paralichthysolivaceus)[J]. Fish & shellfish immunology, 2011, 31(1): 58-65.
[4] Li J,Mo Z,Li G,et al.Generation and evaluation of virulence attenuated mutants ofEdwardsiellatardaas vaccine candidates to combat edwardsiellosis in flounder (Paralichthysolivaceus)[J]. Fish & shellfish immunology, 2015, 43(1): 175-180.
[5] Wang C, Hu Y, Chi H, et al. The major fimbrial subunit protein ofEdwardsiellatarda: vaccine potential, adjuvant effect, and involvement in host infection[J]. Fish & shellfish immunology, 2013, 35(3): 858-865.
[6] Song M, Xie J, Peng X, et al. Identification of protective immunogens from extracellular secretome ofEdwardsiellatarda[J]. Fish & shellfish immunology, 2013, 35(6): 1932-1936.
[7] Yu J E, Yoo A Y, Choi K H, et al. Identification of antigenicEdwardsiellatardasurface proteins and their role in pathogenesis[J]. Fish & shellfish immunology, 2013, 34(2): 673-682.
[8] Li M, Hu Y, Zheng W, et al. Inv1: anEdwardsiellatardainvasinand a protective immunogen that is required for host infection[J]. Fish & shellfish immunology, 2012, 32(4): 586-592.
[9] Zhang M, Wu H, Li X, et al.Edwardsiellatardaflagellar protein FlgD: a protective immunogen against edwardsiellosis[J]. Vaccine, 2012, 30(26): 3849-3856.
[10]Kawai K, Liu Y, Ohnishi K, et al. A conserved 37 kDa outer membrane protein ofEdwardsiellatardais an effective vaccine candidate[J]. Vaccine, 2004, 22(25): 3411-3418.
[11]Plant K P, LaPatra S E. Advances in fish vaccine delivery[J]. Developmental &comparative immunology, 2011, 35(12): 1256-1262.
[12]李新华,沈锦玉,潘晓艺,等. 鱼类口服免疫的研究进展[J]. 水产科学,2010, 29(1): 51-56.
[13]Ferino F, Chauvat F. A promoter-probe vector-host system for the cyanobacterium,SynechocystisPCC6803[J]. Gene, 1989, 84(2): 257-266.
[14]Siripornadulsil S, Dabrowski K, Sayre R. Transgenic microalgae as green cell factories [M].New York:Landes Bioscience andSpringer Science+Business Media, 2007: 122-128.
[15]邓元告,侯李君,邓丽珍,等. 对虾白斑病毒 VP28 基因在聚球藻中的表达与分析[J]. 天津科技大学学报,2008, 23(1): 29-32.
[16]Jia X H, Zhang C L, Shi D J, et al. Oral administration ofAnabaena-expressed VP28 for both drug and food against white spot syndrome virus in shrimp[J]. Journal of Applied Phycology, 2015,28(2): 1-9.
[17]Rippka R. Isolation and purification of cyanobacteria[J]. Methods in enzymology, 1988, 167: 3-27.
[18]马学军,舒跃龙,颜子颖,等. 精编分子生物学实验指南(第4版)[M].北京:科学出版社,2005:55-56.
[19]Elhai J, Wolk C P. Conjugal transfer of DNA to cyanobacteria[J]. Methods in enzymology, 1988, 167: 747-754.
[20]童艳,施定基,冉亮,等. 蓝藻质粒 DNA 提取方法的改进[J]. 植物生理学通讯,2006, 42(2): 281-284.
[21]张亚宁,李晓玥,耿晓娜,等. 迟缓爱德华菌 HB01 外膜蛋白OmpS2基因的克隆表达及其免疫原性研究[J]. 细胞与分子免疫学杂志,2011, 27(10): 1075-1078.
[22]Liu Y, Oshima S, Kurohara K, et al. Vaccine efficacy of recombinant GAPDH ofEdwardsiellatardaagainst Edwardsiellosis[J]. Microbiology and immunology, 2005, 49(7): 605-612.
[23]Zheng Y, Xiao Y, Wu H, et al. Different approaches to expressingEdwardsiellatardaantigen GAPDH in attenuatedVibrioanguillarumfor multivalent fish vaccines[J]. Journal of fish diseases, 2012, 35(8): 569-577.
[24]Liang S, Wu H, Liu B, et al. Immune response of turbot (ScophthalmusmaximusL.) to a broad spectrum vaccine candidate, recombinant glyceraldehyde-3-phosphate dehydrogenase ofEdwardsiellatarda[J]. Veterinary immunology and immunopathology, 2012, 150(3): 198-205.
[25]Li X, Wu H, Zhang M, et al. Secreted glyceraldehyde-3-phosphate dehydrogenase as a broad spectrum vaccine candidate against microbial infection in aquaculture[J]. Letters in applied microbiology, 2012, 54(1): 1-9.
[26]Sun Y, Zheng W J, Hu Y H, et al.EdwardsiellatardaEta1, an in vivo-induced antigen that is involved in host infection[J]. Infection and immunity, 2012, 80(8): 2948-2955.
[27]张春莉,施定基. 白斑综合症病毒 (WSSV) 囊膜蛋白VP28 基因的克隆及在蓝藻中表达载体的构建[J]. 海洋科学,2003, 27(2): 72-76.
[28]施定基,冉亮,李艳,等. 丝状体蓝藻高效表达盒和含有该表达盒的载体[P]. 中国,ZL00132268, 2004.
The Expression ofEdwardsiellatardaEta1-L-Gapdh Fusion Protein in Blue Algae (Anabaenasp. PCC7120)
WANG Zhi, ZHANG Ze-feng, WANG Jing-jing, WANG Chun-mei
(Dept.ofBiopharm.Sci.,BeijingUni.ofChineseMed.,Beijing100102)
In order to expressEdwardsiellatardaEta1-L-Gapdh fusion protein in blue algae (Anabaenasp. PCC7120), genome DNA ofE.tardawas purified and was used as a PCR template for the amplification ofeta1 andgapdhgene, two immunogenic genes that can be developed into vaccine againstE.tarda. Then overlap extension PCR was employed to linketa1 andgapdhgene into the target fusion geneeta1-L-gapdh. The target gene was inserted into the twoBamH I sites of an expression vector pRL489 in order to construct an expression vector that can express the target gene in cyanobacteria. The accuracy of the expression vector was confirmed by such methods as plasmid extraction, PCR, restriction enzyme digestion and sequencing. The confirmed vector was transformed into wide-type fishy algaAnabaenasp. PCC7120 using the method of triparental conjugal transferand neomycin sulfate to screen the transgenic cyanobacteria. The selected positive cyanobacterium was confirmed by plasmid extraction and PCR. Finally the expression of the fusion gene was detected at the transcription and translation level using RT-PCR and Western-blot respectively. The results showed and concluded that the expression vector was successfully constructed, and the target gene was transcribed inAnabaenasp. PCC7120 and a fusion protein was also detected. In addition, the relative expression level of the fusion protein was approximately 2.46%.
Edwardsiellatarda;eta1;gapdh;Anabaenasp. PCC7120; triparental conjugal transfer; fusion protein
北京中医药大学自主选题课题(2013-JYBZZ-JS-139)
王智 男,硕士研究生。研究方向为蓝藻基因工程制药。E-mail: 1169454959@qq.com
* 通讯作者。女,教授,博士,硕土生导师。研究方向为蓝藻基因工程制药。Tel: 010-84738646,E-mail: wchunmei@126.com
2015-12-14;
2016-02-03
Q78
A
1005-7021(2016)05-0078-07
10.3969/j.issn.1005-7021.2016.05.014
