Hedgehog信号通路与胰腺癌吉西他滨固有耐药的相关性研究
2016-12-23邵欣宇丁希伟邹晓平
邵欣宇 顾 燊 丁希伟 邹晓平
南京大学医学院附属鼓楼医院消化科(210008)
Hedgehog信号通路与胰腺癌吉西他滨固有耐药的相关性研究
邵欣宇*顾 燊 丁希伟 邹晓平#
南京大学医学院附属鼓楼医院消化科(210008)
背景:吉西他滨是胰腺癌的一线化疗药物,但化疗耐药问题普遍存在并成为胰腺癌化疗失败的主要原因。Hedgehog(Hh)信号通路是胰腺癌发生、发展的核心信号通路之一,其异常活化与多药耐药相关蛋白表达、肿瘤干细胞的维持和自我更新等明确相关。目的:探讨Hh信号通路相关因子表达与胰腺癌吉西他滨固有耐药的相关性。方法:培养人胰腺癌细胞株CFPAC-1和PANC-1,予吉西他滨、Hh信号通路抑制剂GANT61或激动剂purmorphamine处理,或吉西他滨分别与GANT61或purmorphamine联合处理,以real-timePCR和蛋白质印迹法检测Hh信号通路相关因子Shh、Ptch、Smo、Gli1表达,CCK-8实验检测不同处理对细胞增殖活性的影响。结果:与CFPAC-1细胞相比,PANC-1细胞中的Hh信号通路相关因子表达显著增高(P<0.05),对吉西他滨的敏感性相对较低。GANT61可下调PANC-1细胞中的Gli1表达,并增强细胞对吉西他滨的敏感性,而purmorphamine则可上调CFPAC-1细胞中的Gli1表达,并降低细胞对吉西他滨的敏感性,差异均有统计学意义(P<0.05)。结论:Hh信号通路相关因子表达与胰腺癌吉西他滨固有耐药相关,抑制Hh信号通路异常活化可增加胰腺癌细胞对吉西他滨的敏感性。
胰腺肿瘤; 吉西他滨;Hedgehog信号通路;Hedgehog蛋白质类; 抗药性,肿瘤
胰腺癌是恶性程度极高的消化系统肿瘤,2015年发表的肿瘤统计报告显示胰腺癌在美国肿瘤相关死因中居第四位[1];2004—2009年间,上海市胰腺癌发病率为6.7/10万,5年总生存率约4.1%,中位生存期仅为3.9个月[2]。80%以上的胰腺癌患者确诊时已处于肿瘤晚期,无法进行外科干预[3],因此,胰腺癌的综合治疗显得尤为重要。吉西他滨(gemcitabine, 2’,2’-difluoro-2’-deoxycytidine)是胰腺癌的一线化疗药物[4-5],但化疗耐药问题普遍存在并成为胰腺癌化疗失败的主要原因。化疗耐药可分为固有耐药和获得性耐药,前者为细胞在接受化疗前即存在的固有的对药物不敏感,后者指在化疗过程中,原本敏感的细胞通过基因突变以及其他适应性改变而变得耐药。Hedgehog(Hh)信号通路是胰腺癌发生、发展的核心信号通路之一,该信号通路相关蛋白在正常胰腺组织中不表达,但在胰腺癌及其癌前病变中存在异常表达[6]。本研究选择两株对吉西他滨敏感性不同的人胰腺癌细胞株CFPAC-1和PANC-1,通过检测Hh信号通路相关因子Shh、Ptch、Smo、Gli1在两者中的表达情况,以及Hh信号通路抑制剂或激动剂对细胞吉西他滨敏感性的影响,探讨Hh信号通路相关因子表达与胰腺癌吉西他滨固有耐药的相关性。
材料与方法
一、细胞株和主要试剂
人胰腺癌细胞株CFPAC-1、PANC-1购自中国科学院上海生命科学研究院细胞资源中心,送GENEWIZ公司以短串联重复序列(short tandem repeat)图谱分析方法进行鉴定,结果与细胞库和文献报道相符。两株细胞均按贴壁方法培养于含10%胎牛血清的DMEM培养基中,5% CO2、37 ℃恒温培养箱内培养。细胞生长至接近融合状态时,以含EDTA 的0.25% 胰酶消化传代,2~3 d一次,取对数生长期细胞用于实验。
吉西他滨购自中国食品药品检定研究院,溶于双蒸水中配制成2.5 mmol/L的母液备用; GANT61、purmorphamine购自Sigma-Aldrich Co. LLC.,溶于DMSO中配制成10 mmol/L的母液备用。
TRIzol®RNA分离试剂(Thermo Fisher Scientific Inc.),PrimeScriptTMRT reagent Kit (Perfect Real Time)、SYBR®Premix Ex TaqTMⅡ(Tli RNaseH Plus)(TAKARA BIO INC.),real-time PCR引物由生工生物工程(上海)股份有限公司合成。Shh: F 5’-GCT CGG TGA AAG CAG AGA AC-3’, R 5’-CCA GGA AAG TGA GGA AGT CG-3’; Ptch: F 5’-GGA GCA GAT TTC CAA GGG GA-3’, R 5’-CCA CAA CCA AGA ACT TGC CG-3’; Smo: F 5’-GCT ACA ACG TGT GCC TGG G-3’, R 5’-CAT TCC GGA GGC CCG AC-3’; Gli1: F 5’-CCC AGA CAG AGG CCC ACT C-3’, R 5’-AGA TGT GCA TCG CGA GTT GA-3’; β-actin: F 5’-CCT GGC ACC CAG CAC AAT-3’, R 5’-AGT ACT CCG TGT GGA TCG GC-3’。Shh兔多克隆抗体、Ptch羊多克隆抗体、Gli1兔多克隆抗体(Santa Cruz Biotechnology),Smo兔多克隆抗体(Abcam plc.)。CCK-8细胞增殖/细胞毒性检测试剂盒[东仁化学科技(上海)有限公司]。
二、方法
1. Real-time PCR:取对数生长期CFPAC-1、PANC-1细胞,按实验要求进行不同处理。48 h或72 h后收集细胞,预冷PBS洗涤2次,TRIzol®试剂提取总RNA,逆转录合成cDNA,行real-time PCR,操作步骤参照试剂盒说明书。2-△△Ct法计算目的基因mRNA相对表达量。每组设3个复孔,每次实验至少重复3次。
2. 蛋白质印迹法:取对数生长期CFPAC-1、PANC-1细胞,按实验要求进行不同处理。48 h或72 h后收集细胞,预冷PBS洗涤2次,加入蛋白裂解液冰上裂解,4 ℃ 12 000 r/min离心20 min,取上清,BCA法蛋白定量。取30 μg总蛋白上样,SDS-PAGE电泳分离1~2 h,蛋白转移至PVDF膜,5%牛奶封闭2 h,加入一抗4 ℃孵育过夜,TBST洗涤,加入二抗孵育2 h,TBST洗涤,ECL法自动曝光成像。实验重复3次。
3. CCK-8实验:取对数生长期CFPAC-1、PANC-1细胞接种于96孔板,24 h后按实验要求进行不同处理,CFPAC-1细胞培养48 h,PANC-1细胞培养 72 h,吸去培养基,每孔加入10 μL CCK-8与90 μL完全培养基混合物,2 h后酶标仪测定450 nm波长处吸光度值(A值)。细胞活力=(A待测组-A空白组)/(A对照组-A空白组)×100%。
三、统计学分析
结 果
一、Hh信号通路相关因子在CFPAC-1、PANC-1细胞中呈差异性表达
既往文献报道Hh信号通路相关因子在胰腺癌组织中表达异常[6]。本实验分别在基因和蛋白水平检测了该通路中的主要因子Shh、Ptch、Smo、Gli1在人胰腺癌细胞株CFPAC-1、PANC-1中的表达情况,结果显示两株细胞中的Hh信号通路均异常活化,相关因子尤其是转录因子Gli1在PANC-1细胞中的表达显著高于CFPAC-1细胞(P<0.05)(图1)。
二、PANC-1细胞对吉西他滨相对不敏感
经不同浓度吉西他滨处理后(CFPAC-1细胞48h,PANC-1细胞72h),低倍镜下可见贴壁细胞减少,漂浮细胞增多,高倍镜下可见贴壁细胞形态改变,伸出较多突起,细胞内可见空泡。CCK-8实验显示,吉西他滨浓度越高,细胞增殖抑制作用越明显,CFPAC-1细胞50%抑制浓度(IC50)为101nmol/L,PANC-1细胞IC50为5.346mmol/L,CFPAC-1细胞对吉西他滨的敏感性明显高于PANC-1细胞(图2)。Hh信号通路相关因子表达较高的PANC-1细胞对吉西他滨相对不敏感,提示Hh信号通路异常活化可能与胰腺癌细胞的固有耐药相关。
三、抑制Gli1可增加PANC-1细胞对吉西他滨的敏感性
GANT61为Hh信号通路抑制剂,主要作用为干扰转录因子Gli1与DNA结合,从而抑制其靶基因转录[7]。由于Gli1能调控自身转录,因此GANT61有抑制Gli1合成的功能。以GANT61处理Gli1表达较高的PANC-1细胞72h,real-timePCR和蛋白质印迹法检测显示GANT61可从基因和蛋白水平下调Gli1表达(P<0.05)(图3)。CCK-8实验显示,GANT61处理72h可抑制PANC-1细胞的增殖活性(图4A),选用对增殖活性影响较小的5μmol/LGANT61联合吉西他滨处理PANC-1细胞72h,增殖抑制作用较吉西他滨单药处理增强,组间差异有统计学意义(P<0.05)(图4B),且此差异大于5μmol/LGANT61单独作用的效果,表明抑制Gli1可增强PANC-1细胞对吉西他滨的敏感性。
四、激活Smo可上调CFPAC-1细胞的Gli1表达并降低细胞对吉西他滨的敏感性
Purmorphamine能选择性结合并激活Smo,从而激活Hh信号通路[8]。以purmorphamine处理Gli1表达较低的CFPAC-1细胞48h,real-timePCR和蛋白质印迹法检测显示purmorphamine可从基因和蛋白水平上调Gli1表达(P<0.05)(图5)。CCK-8实验显示,purmorphamine处理48h可增强CFPAC-1细胞的增殖活性(图6A),选用对增殖活性影响较小的5μmol/Lpurmorphamine联合吉西他滨处理CFPAC-1细胞48h,发现purmorphamine可减弱吉西他滨单药处理的增殖抑制作用,组间差异有统计学意义(P<0.05)(图6B),且此差异大于5μmol/Lpurmorphamine单独作用的效果,表明激活Smo可降低CFPAC-1细胞对吉西他滨的敏感性。
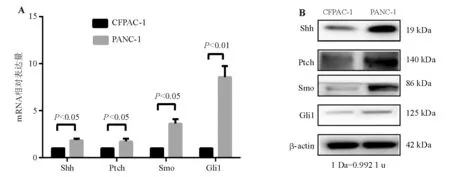
A:real-time PCR;B:蛋白质印迹法
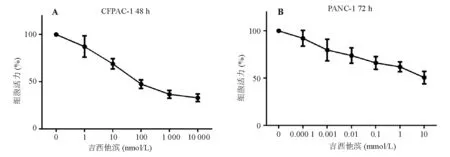
图2 吉西他滨抑制CFPAC-1、PANC-1细胞的增殖活性(CCK-8实验)
讨 论
Hh信号通路在哺乳动物的胚胎发育过程中起重要作用,出生后,该通路在正常情况下几乎无活性,其异常活化与多种恶性肿瘤的发生、生长、侵袭、 转移等恶性生物学行为有关。在哺乳动物中,Hh信号通路主要有3个配体蛋白:SonicHedgehog(Shh)、DesertHedgehog(Dhh)和IndianHedgehog(Ihh)。其激活途径为:由自分泌或旁分泌途径产生的hh(包括Shh、Dhh、Ihh)作用于肿瘤抑制因子Patched(Ptch,一种十二次跨膜受体),两者结合后,释放原先被Ptch抑制的七次跨膜受体Smoothened(Smo),Smo移动至初级纤毛,使肿瘤抑制因子sufu(suppressoroffused)与转录因子Gli(包括Gli1、Gli2和Gli3)解聚,Gli进入细胞核,调节靶基因转录,其靶基因包括Gli1、Ptch以及多种促进细胞增殖、存活的分子如Myc、Bcl-2、cyclin等。其他如Ptch基因突变、sufu蛋白缺失、Gli过表达等亦可激活Hh信号通路。一项针对胰腺癌患者的全基因组分析显示,几乎所有患者均存在Hh信号通路基因突变[9]。在正常胰腺组织、胰腺上皮内瘤变至胰腺癌的进展过程中,Hh信号通路相关蛋白的表达水平逐渐增高[6,10],癌组织中相关蛋白Shh、Gli1表达水平越高,患者预后越差[11]。因此,针对Hh信号通路的研究在胰腺癌研究领域中居重要地位。

A:real-time PCR;B:蛋白质印迹法
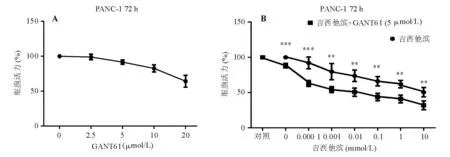
与同浓度吉西他滨单药处理比较,**P<0.01,***P<0.001
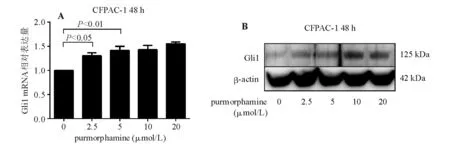
A:real-time PCR;B:蛋白质印迹法
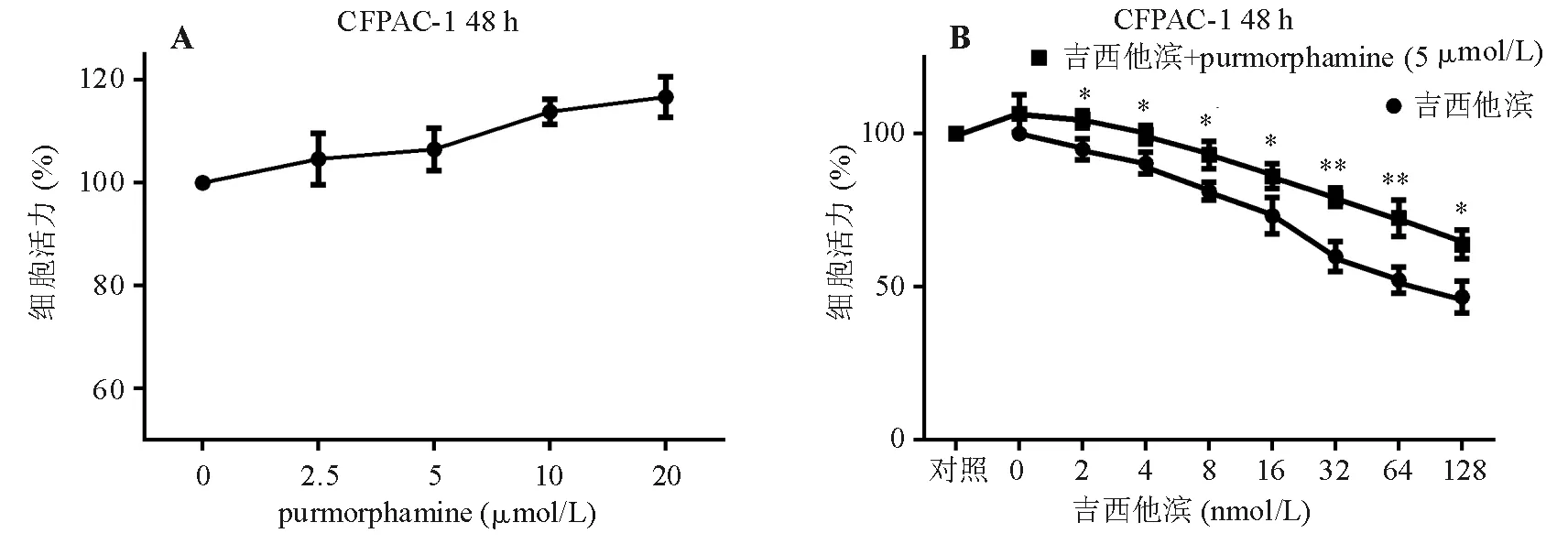
与同浓度吉西他滨单药处理比较,*P<0.05,**P<0.01
吉西他滨是胰腺癌的一线化疗药物之一[4-5],但耐药现象限制了该药的临床应用。探索胰腺癌对吉西他滨耐药的机制可为胰腺癌的有效治疗、精准治疗提供理论依据。胰腺癌治疗困难的原因之一为癌组织中血管相对较少,使药物难以被转运、吸收。近年众多研究证实Hh信号通路可影响胰腺癌间质的形成和分化,抑制该信号通路可减少肿瘤相关间质组织,增加癌组织中的血管密度,从而增加药物灌注[12-13]。肿瘤干细胞的存在亦为影响胰腺癌吉西他滨化疗效果的重要因素[14-15]。研究表明Hh信号通路在肿瘤干细胞的维持和自我更新方面起重要作用[16-17]。此外,Hh信号通路还与多药耐药相关蛋白ABCG2(BCRP)、ABCB1(MDR1)等明确相关[18-20],可介导肿瘤细胞的多药耐药。上述发现为提出Hh信号通路与胰腺癌对吉西他滨的耐药程度存在相关性的假设奠定了理论基础。
本研究检测了Hh信号通路相关因子在两株人胰腺癌细胞株CFPAC-1、PANC-1中的表达情况,发现PANC-1细胞中Shh、Ptch、Smo、Gli1在基因和蛋白水平的表达均显著高于CFPAC-1细胞,尤其是Gli1,CCK-8实验则显示吉西他滨对PANC-1细胞的IC50远高于CFPAC-1细胞,提示Hh信号通路异常活化可能降低胰腺癌细胞对吉西他滨的敏感性。为进一步证实此推测,本研究分别将Hh信号通路抑制剂和激动剂与吉西他滨联合应用,检测两者对胰腺癌细胞吉西他滨敏感性的影响。结果显示,在Gli1表达较高的PANC-1细胞中,Gli1抑制剂GANT61处理可使细胞对吉西他滨的敏感性显著增强;而在Gli1表达较低的CFPAC-1细胞中,Smo激动剂purmorphamine处理可上调Gli1表达并显著降低细胞对吉西他滨的敏感性。上述结果证实了之前的推测:Hh信号通路异常活化与胰腺癌吉西他滨固有耐药相关。这一结论为胰腺癌患者的精准治疗提供了可能的靶点,但仍需开展相关临床研究。
尽管针对Hh信号通路的特异性小分子抑制剂GDC-0449(vismodegib)在临床试验中显示出较好的抗肿瘤活性[21-22],但在胰腺癌的治疗中却未显示出能增强吉西他滨的治疗效果[23]。可能原因之一是GDC-0449为Smo抑制剂,而Smo为Gli1的上游分子,Hh信号通路受多个旁系通路影响,下游Gli1表达水平是多种方式综合调控的结果,尚存在一些其他信号通路如PI3K/AKT、RAS/RAF/MEK/ERK轴、Wnt等,可通过非Smo依赖性途径调节Gli1表达[24-25]。由此推测在Gli1水平抑制Hh信号通路产生的抗肿瘤效应应优于Smo抑制剂。因此本研究选择Gli1特异性阻断剂GANT61作为Hh信号通路抑制剂,发现经GANT61处理后,原先对吉西他滨相对不敏感的PANC-1细胞敏感性显著增强。
综上所述,Hh信号通路相关因子表达与胰腺癌吉西他滨固有耐药相关,抑制Hh信号通路异常活化可增加胰腺癌细胞对吉西他滨的敏感性。这一发现或许能为未来解决胰腺癌吉西他滨耐药问题提供一个新的思路。
1SiegelRL,MillerKD,JemalA.Cancerstatistics, 2015[J].CACancerJClin, 2015, 65 (1): 5-29.
2LuoJ,XiaoL,WuC,etal.Theincidenceandsurvivalrateofpopulation-basedpancreaticcancerpatients:ShanghaiCancerRegistry2004-2009[J].PLoSOne, 2013, 8 (10):e76052.
3VincentA,HermanJ,SchulickR,etal.Pancreaticcancer[J].Lancet, 2011, 378 (9791): 607-620.
4TemperoMA,ArnolettiJP,BehrmanSW,etal;NationalComprehensiveCancerNetworks.PancreaticAdenocarcin-oma,version2.2012:featuredupdatestotheNCCNGuidelines[J].JNatlComprCancNetw, 2012, 10 (6): 703-713.
5deSousaCavalcanteL,MonteiroG.Gemcitabine:metabolismandmolecularmechanismsofaction,sensitivityandchemoresistanceinpancreaticcancer[J].EurJPharmacol, 2014, 741: 8-16.
6ThayerSP,diMaglianoMP,HeiserPW,etal.Hedgehogisanearlyandlatemediatorofpancreaticcancertumorigenesis[J].Nature, 2003, 425 (6960): 851-856.
7LauthM,BergströmA,ShimokawaT,etal.InhibitionofGLI-mediatedtranscriptionandtumorcellgrowthbysmall-moleculeantagonists[J].ProcNatlAcadSciUSA, 2007, 104 (20): 8455-8460.
8SinhaS,ChenJK.PurmorphamineactivatestheHedgehogpathwaybytargetingSmoothened[J].NatChemBiol, 2006, 2 (1): 29-30.
9JonesS,ZhangX,ParsonsDW,etal.Coresignalingpathwaysinhumanpancreaticcancersrevealedbyglobalgenomicanalyses[J].Science, 2008, 321 (5897): 1801-1806.
10BardeesyN,DePinhoRA.Pancreaticcancerbiologyandgenetics[J].NatRevCancer, 2002, 2 (12): 897-909.
11MaréchalR,BachetJB,CalommeA,etal.SonichedgehogandGli1expressionpredictoutcomeinresectedpancreaticadenocarcinoma[J].ClinCancerRes, 2015, 21 (5): 1215-1224.
12NeesseA,MichlP,FreseKK,etal.Stromalbiologyandtherapyinpancreaticcancer[J].Gut, 2011, 60 (6): 861-868.
13OliveKP,JacobetzMA,DavidsonCJ,etal.InhibitionofHedgehogsignalingenhancesdeliveryofchemotherapyinamousemodelofpancreaticcancer[J].Science, 2009, 324 (5933): 1457-1461.
14HongSP,WenJ,BangS,etal.CD44-positivecellsareresponsibleforgemcitabineresistanceinpancreaticcancercells[J].IntJCancer, 2009, 125 (10): 2323-2331.
15ShahAN,SummyJM,ZhangJ,etal.Developmentandcharacterizationofgemcitabine-resistantpancreatictumorcells[J].AnnSurgOncol, 2007, 14 (12): 3629-3637.
16ClementV,SanchezP,deTriboletN,etal.HEDGEHOG-GLI1signalingregulateshumangliomagrowth,cancerstemcellself-renewal,andtumorigenicity[J].CurrBiol, 2007, 17 (2): 165-172.
17LiuS,DontuG,MantleID,etal.HedgehogsignalingandBmi-1regulateself-renewalofnormalandmalignanthumanmammarystemcells[J].CancerRes, 2006, 66 (12): 6063-6071.
18SinghRR,KunkallaK,QuC,etal.ABCG2isadirecttranscriptionaltargetofhedgehogsignalingandinvolvedinstroma-induceddrugtoleranceindiffuselargeB-celllymphoma[J].Oncogene, 2011, 30 (49): 4874-4886.
19ChenY,BieberMM,TengNN.HedgehogsignalingregulatesdrugsensitivitybytargetingABCtransportersABCB1andABCG2inepithelialovariancancer[J].MolCarcinog, 2014, 53 (8): 625-634.
20Sims-MourtadaJ,IzzoJG,AjaniJ,etal.SonicHedgehogpromotesmultipledrugresistancebyregulationofdrugtransport[J].Oncogene, 2007, 26 (38): 5674-5679.
21LoRussoPM,RudinCM,ReddyJC,etal.PhaseⅠtrialofhedgehogpathwayinhibitorvismodegib(GDC-0449)inpatientswithrefractory,locallyadvancedormetastaticsolidtumors[J].ClinCancerRes, 2011, 17 (8): 2502-2511.
22SekulicA,MigdenMR,OroAE,etal.Efficacyandsafetyofvismodegibinadvancedbasal-cellcarcinoma[J].NEnglJMed, 2012, 366 (23): 2171-2179.
23KimEJ,SahaiV,AbelEV,etal.PilotclinicaltrialofhedgehogpathwayinhibitorGDC-0449 (vismodegib)incombinationwithgemcitabineinpatientswithmetastaticpancreaticadenocarcinoma[J].ClinCancerRes, 2014, 20 (23): 5937-5945.
24InghamPW,NakanoY,SegerC.MechanismsandfunctionsofHedgehogsignallingacrossthemetazoan[J].NatRevGenet, 2011, 12 (6): 393-406.
25AbergerF,KernD,GreilR,etal.CanonicalandnoncanonicalHedgehog/GLIsignalinginhematologicalmalignancies[J].VitamHorm, 2012, 88: 25-54.
(2016-03-29收稿;2016-04-25修回)
Hedgehog Signaling Pathway Mediates Gemcitabine Intrinsic Chemoresistance in Pancreatic Cancer
SHAOXinyu,GUShen,DINGXiwei,ZOUXiaoping.
DepartmentofGastroenterology,theAffiliatedDrumTowerHospitalofNanjingUniversityMedicalSchool,Nanjing(210008)
Correspondence to: ZOU Xiaoping, Email: 13770771661@163.com
Background: Gemcitabine is the standard first-line chemotherapy for pancreatic cancer. However, chemoresistance commonly occurs and is the main cause of therapeutic failure. Hedgehog (Hh) pathway is a core signaling pathway in the initiation and progression of pancreatic cancer, the aberrant activation of Hh pathway is associated with the expression of multidrug resistance related proteins and the maintenance and self-renewal of cancer stem cells. Aims: To investigate the correlation between expressions of Hh pathway related factors and gemcitabine intrinsic chemoresistance in pancreatic cancer. Methods: Human pancreatic cancer cell line CFPAC-1 and PANC-1 were treated with gemcitabine, GANT61 -- the inhibitor of Hh pathway, purmorphamine -- the agonist of Hh pathway, and gemcitabine combined with GANT61 or purmorphamine, respectively. Expressions of the Hh pathway related factors Shh, Ptch, Smo and Gli1 were detected by real-time PCR and Western blotting, and CCK-8 assay was used to assess the effect of various interventions on cell viability. Results: Compared with CFPAC-1 cells, expressions of Hh pathway related factors were significantly higher in PANC-1 cells (P<0.05) and PANC-1 cells was more resistant to gemcitabine. GANT61 could down-regulate Gli1 expression and increase the sensitivity to gemcitabine in PANC-1 cells, whereas purmorphamine could up-regulate the expression of Glil and decrease the sensitivity to gemcitabine in CFPAC-1 cells (Pall <0.05). Conclusions: Expressions of Hh pathway related factors are correlated with gemcitabine intrinsic chemoresistance in pancreatic cancer. Inhibiting Hh pathway aberrant activation might increase the sensitivity of pancreatic cancer cells to gemcitabine.
Pancreatic Neoplasms; Gemcitabine; Hedgehog Pathway; Hedgehog Proteins; Drug Resistance, Neoplasm
10.3969/j.issn.1008-7125.2016.11.004
*Email:xinxinyuxin0711@163.com
#本文通信作者,Email: 13770771661@163.com
