正交试验优选乌药中去甲异波尔定的提取工艺
2016-12-21刘译
刘译
(湖南省怀化市食品药品检验所,湖南 怀化 418000)
正交试验优选乌药中去甲异波尔定的提取工艺
刘译
(湖南省怀化市食品药品检验所,湖南 怀化 418000)
目的 优选乌药中去甲异波尔定的提取工艺。方法 采用正交试验设计,以去甲异波尔定提取率为考察指标,考察提取前浸泡时间、提取溶剂、提取方法、固液比、提取时间、提取温度等因素,同时测定干浸膏收率。结果 最佳方案为提取前浸泡20 min,60%的乙醇水溶液提取溶剂,最终确定提取方法为超声提取、固液比1∶12、提取时间为20 min、提取温度为45℃。结论 该方法简便、稳定、可行,可有效提取乌药中去甲异波尔定的含量,为以乌药为原料的抗宫炎系列药品提供质量控制依据。
乌药;去甲异波尔定;提取工艺;正交设计;高效液相色谱法
乌药来源于樟科植物乌药 Lauraceae Lindera.aggregata(Sims)Kosterm.的干燥块根,首载于《本草拾遗》,具有行气止痛、温肾散寒的功能,用于寒凝气滞、胸腹胀痛、气逆喘急、膀胱虚冷、遗尿尿频、疝气疼痛、经寒腹痛[1]。乌药主产于浙江、湖南、安徽、江西等省,以浙江天台所产乌药品质最佳。乌药主要含挥发油、异喹啉类生物碱、黄酮类及呋喃倍半萜类成分。早期乌药的质量研究主要集中在乌药内酯或乌药醚内酯等脂溶性成分的含量测定,后来发现乌药中异喹啉类生物碱如去甲异波尔定等水溶性成分也具有一定药理活性。近年来,逐渐将乌药生物碱纳入质量研究体系,2015年版《中国药典(一部)》乌药质量标准增加了去甲异波尔定含量测定,规定去甲异波尔定不得少于0.40%。传统乌药使用多以水煎煮应用于临床,乌药干浸膏也为水煎煮提取制成的浸膏[2]。乌药醚内酯易溶于有机溶剂,去甲异波尔定为异喹啉类生物碱,结构中既含有氮原子也有羟基,属于既溶于有机溶剂又溶于水的化合物。已有乌药中乌药醚内酯提取工艺[3]的相关报道,但关于从乌药中提取去甲异波尔定的文献报道不多。本研究中以去甲异波尔定提取率为考察指标,优选了乌药中去甲异波尔定的提取工艺参数,也为以乌药为原料之一的抗宫炎系列药品质量控制提供依据。现报道如下。
1 仪器与试药
1.1 仪器
LC-20AT型及SPD-20A型高效液相色谱仪,包括岛津四元梯度泵、在线脱气机、自动进样器、柱温箱、SPD检测器、LCsolution色谱工作站;TOLEDO XSE105型电子分析天平(Mettler公司);HHS型电热恒温水浴锅(上海博讯实业有限公司);CBL9960A型超声波清洗器(天津科贝尔光电技术有限责任公司);202-1A型电热恒温干燥箱及FW100微型粉碎机(天津市泰斯特仪器有限公司)。
1.2 试药
乌药购于湖南邵东县廉桥中药材专业市场,经怀化市食品药品检验所唐勇主任药师鉴定为樟科山胡椒属植物乌药 Lauraceae Lindera.aggregata(Sims)Kosterm.的干燥块根;去甲异波尔定对照品(纯度为95.7%,中国食品药品检定研究院,批号为111825-201402);甲醇、乙腈、甲酸、三乙胺为色谱纯;水为二次重蒸馏水,其他试剂均为分析纯。
2 方法与结果
2.1 去甲异波尔定含量测定
2.1.1 色谱条件[2]与系统适用性试验
色谱柱:Zorbax SB C18柱(250 mm×4.6 mm,5 μm);流动相:乙腈(A)-0.5%甲酸和0.1% 三乙胺的水溶液(B),梯度洗脱,0~13 min时流动相A从10%→22%,13~22 min时流动相A维持22%);流速:1.0 mL/min;检测波长:280 nm;进样量5 μL。理论板数按去甲异波尔定峰计算应大于5 000。在此条件下,色谱图见图1。
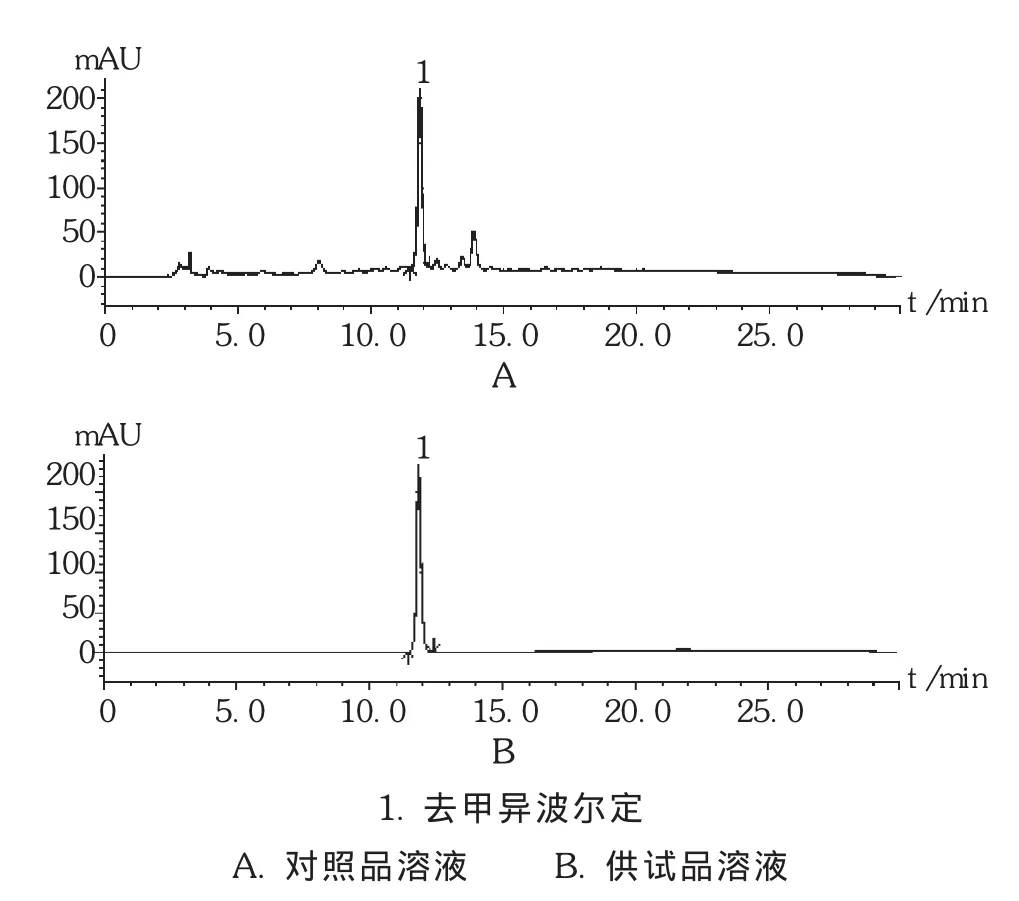
图1 高效液相色谱图
2.1.2 溶液制备
对照品溶液:精密称取去甲异波尔定对照品适量,加甲醇-0.5%盐酸的混合溶液(2∶1)制成每1 mL中含去甲异波尔0.2 mg,摇匀,即得。
供试品溶液:取本品粉末0.5 g,精密称定,置250 mL圆底烧瓶中,精密加入甲醇-稀盐酸(0.5%)的混合液(2∶1)25 mL,称定质量,加热回流并保持微沸1 h,放冷,再称定质量,用甲醇-稀盐酸(0.5%)的混合液(2∶1)补足减失的质量,摇匀,静置,取上清液,0.45 μm滤膜过滤即得。
2.1.3 方法学考察
线性关系考察:取对照品溶液,分别以1.0,2.0,5.0,8.0,10.0,12.0,20.0 μL进样分析。按拟订色谱条件测定峰面积,以峰面积积分值(Y)为纵坐标、对照品进样量(X)为横坐标作图,进行线性回归。得回归方程Y=2 779 017.373X-82 361.461,r=0.999 49(n=7)。结果表明,去甲异波尔质量浓度在0.202 8~4.056 0 g/L范围内与峰面积积分值呈良好线性关系。
精密度试验:精密吸取同一对照品溶液5 μL,注入液相色谱仪,连续进样6次。结果的 RSD为0.13%(n=6),表明仪器精密度良好。
稳定性试验:取同一供试品溶液,分别于0,2,4,6,10,18,24,48 h时连续进样3次,各5 μL,测定去甲异波尔定峰面积。结果的 RSD为0.54%(n=8),表明供试品溶液在48 h内基本稳定。
重复性试验:取同一批乌药药材,依法制备6份供试品溶液,精密吸取5 μL注入液相色谱仪进行测定。结果的 RSD为1.57%(n=6),表明该方法重复性良好。
加样回收试验:取已知含量的供试品6份,分别精密加入一定量的去甲异波尔定对照品,照供试品溶液制备方法制备溶液,精密吸取5 μL注入色谱仪中,测定峰面积,计算回收率。结果表明,6次加样回收率均在95%~105%,平均值99.34%,RSD为1.44%(n=6)。
2.2 干浸膏收率的测定
取各试验所得的浸膏约1 g,精密称定,水浴蒸干后,于105℃干燥5 h,置干燥器中冷却30 min,迅速精密称定质量,再于105℃干燥1 h,冷却称定质量,至两次质量的差异不超过5 mg为止,记录干浸膏质量,计算干浸膏收率。
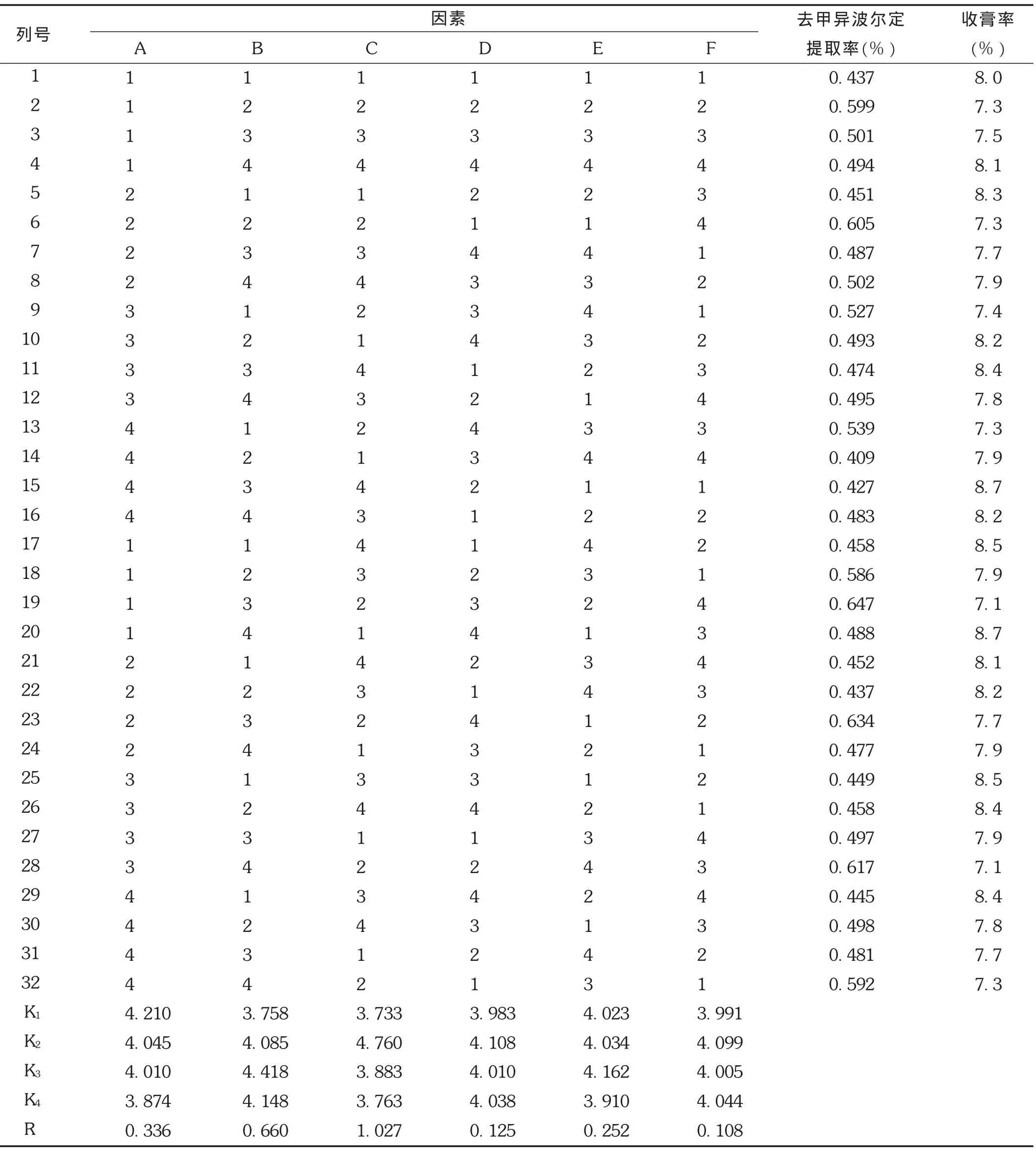
表2 正交试验设计与结果
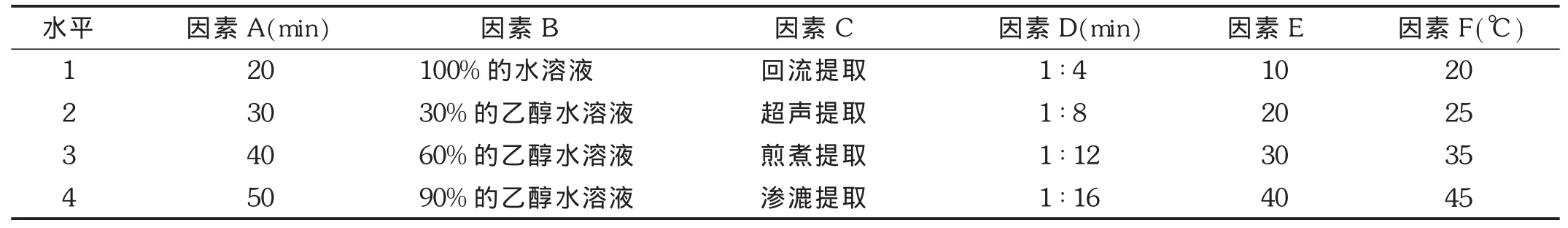
表1 L25(46)正交试验因素水平表
2.3 提取工艺正交试验
2.3.1 正交试验
以去甲异波尔定提取率为考察指标,采用 L25(46)正交表进行试验,考察提取前浸泡时间(因素A)、提取溶剂(因素B)、提取方法(因素C)、固液比(因素D)、提取时间(因素E)、提取温度(因素F)6个因素。水平因素表和试验结果见表1和表2,方差分析见表3。
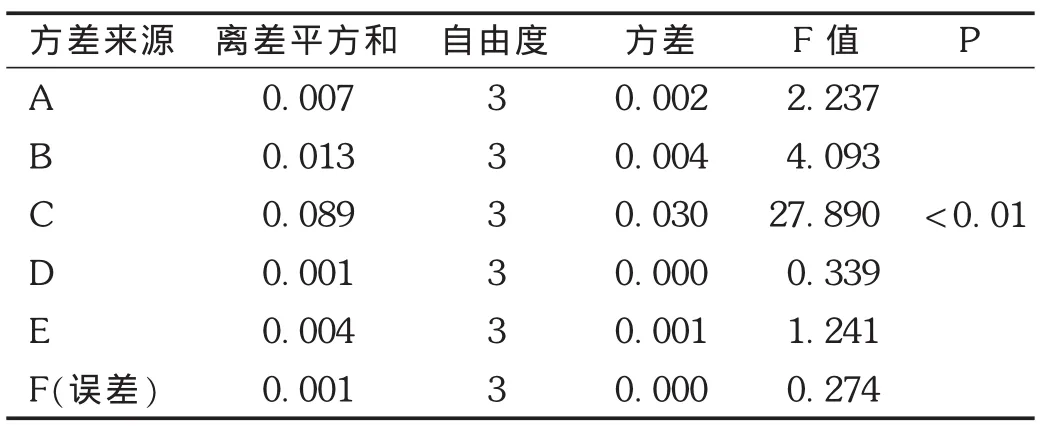
表3 方差分析结果
对去甲异波尔定的提取总量(以原药材计)进行分析,由表2及表3可知,影响去甲异波尔定提取率的各因素主次顺序为C>B>A>E>D>F。其中,因素C的影响具有显著性意义(P<0.01),因素 B有一定影响(P<0.1),因素A,E,D,F的影响无显著性,故确定最佳提取方案为A1B3C2D3E2F4,即提取方法为超声提取、固液比1∶12、提取时间为20 min、提取温度为45℃。在去甲异波尔定的提取率相同的情况下,收膏率越低越好,对收膏率进行直观分析所得最佳结果也为A1B3C2D3E2F4。因此,以 A1B3C2D3E2F4为最佳提取工艺方案。
2.3.2 验证试验
以A1B3C2D3E2F4方案进行3次验证试验,结果见表4。各项指标的 RSD均<1.8%,去甲异波尔定的提取率较高,而收膏率较低,表明正交试验所筛选的工艺稳定可行。
3 讨论
3.1 提取方法选择
由方差分析可知,影响提取效率最大的因素为提取方法。本试验中通过超声提取这一物理过程,在整个浸提过程中基本无化学反应发生,这与超声提取能产生并传递强大的能量的空化作用,并能产生许多次级作用有关[4-5]。超声提取避免了常规回流法、煎煮法等长时间加热对乌药中去甲异波尔定的的不良影响,与传统方法比较缩短了提取时间,提高了提取率。
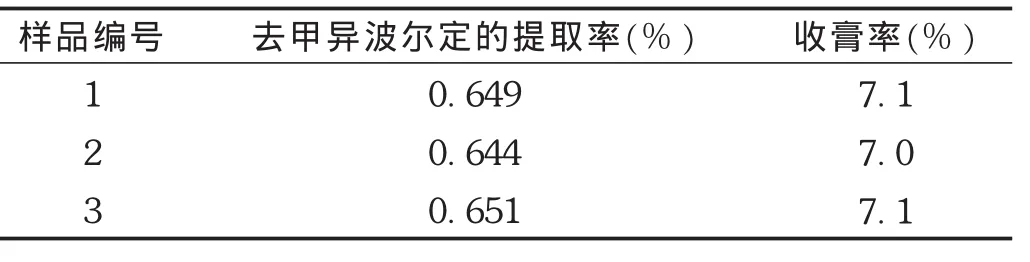
表4 正交试验最佳提取工艺验证试验结果
3.2 提取溶剂考察
提取溶剂及浓度的选择对结果也有一定影响,用含水的乙醇提取比用纯水提取的提取率更高。60%的乙醇水溶液提取去甲异波尔定比用30%乙醇水溶液的提取率要高,而和90%乙醇的提取率差不多,故选用60%的乙醇水溶液。
3.3 提取工艺确定
通过正交试验得到乌药中去甲异波尔定的最佳提取工艺:提取前浸泡时间为20 min,提取溶剂为60%的乙醇水溶液,提取方法为超声提取,固液比1∶12,提取时间为20 min,提取温度为45℃。该方法简单、稳定、可靠,可作为乌药提取去甲异波尔定的参考标准,也可为以乌药为原料之一的抗宫炎系列药品质量控制提供依据。
[1]国家药典委员会.中华人民共和国药典(一部)[M].北京:中国医药科技出版社,2015:77.
[2]姚新生.天然药物化学[M].第5版.北京:人民卫生出版社,2003:20.
[3]陈方亮,余翠琴,徐仙娥.正交试验优选乌药的提取工艺[J].中草药,2009,40(7):1 079-1 081.
[4]徐天生,梁 燕,江莉华,等.用正交试验法研究超声提取丹参多糖的最佳工艺[J].中成药,2008,30(2):270-271.
[5]杨祖金,钟 理,谢小霞,等.超声波法强化提取罗汉果皂甙的工艺研究[J].中药材,2007,30(8):1 019-1 021.
Optimization of Extract Technology of Norisoboldine from Radix Linderae by Orthogonal Experiment
Liu Yi
(Huaihua Institute for Food and Drug Contro,Huaihua,Hunan,China 418000)
Objective Tooptimize theoptionalextraction method of norisoboldine from Radix Linderae.Methods Thecontent of norisoboldine was used as index to evaluate the technologies based on orthogonal design,considering ointment rate at the same time,in which the 6 factors were the soaking time before extraction,extraction solvent,extraction method,ratio of solid to liquid,extraction time, and extracting temperature.Results The optimum extraction processes was soaking for 20 min before extraction,60% ethanol,ultrasonic extraction technique,solid-liquid ratio 1∶12,the extraction time was 20 min,the extracting temperature was 45℃.Conclusion The method is simple,stable and feasible.
Radix Linderae;norisoboldine;extraction process;orthogonal experiment;HPLC
R284.1;R282.71
A
1006-4931(2016)21-0010-04
刘译(1964-),男,副主任药师,主要从事中药研发、检验和检测,(电子信箱)hhsyjs@126.com。
2016-07-13)
湖南省食品药品监督管理局2015年度食品药品安全科技重点项目,项目编号:湘食药科R201501。
