卵泡期和黄体期山羊垂体miRNAs表达谱及差异分析
2016-12-13凌英会睢梦华张运海章孝荣
凌英会,朱 龙,睢梦华,郑 琪,吴 昊,张运海,章孝荣,3*
(1.安徽农业大学动物科技学院,合肥 230036; 2.安徽地方畜禽遗传资源保护与生物育种省级实验室,合肥 230036;3.安徽省羊繁育工程技术研究中心,合肥 230036)
卵泡期和黄体期山羊垂体miRNAs表达谱及差异分析
凌英会1,2,朱 龙1,2,睢梦华1,2,郑 琪1,2,吴 昊1,2,张运海1,2,章孝荣1,2,3*
(1.安徽农业大学动物科技学院,合肥 230036; 2.安徽地方畜禽遗传资源保护与生物育种省级实验室,合肥 230036;3.安徽省羊繁育工程技术研究中心,合肥 230036)
旨在通过高通量测序的方法筛选与分析卵泡期和黄体期安淮山羊垂体中差异表达miRNAs,探索垂体在转录后水平调节山羊从卵泡期-黄体期过渡中发挥的作用。测序结果表明,卵泡期和黄体期文库分别得到11 319 069和11 432 846条原始序列。经过去除杂质后,得到11 162 888和11 016 138条纯净序列。在两个不同时期的文库中,分别检测到148和150种差异表达的miRNA,有147种miRNA共同表达;分别预测到52和95种差异表达的miRNA,有27种miRNA两文库共同表达。已知miRNA中,let-7f在两文库表达量最高,且差异极显著;新miRNA中,novel-miR-159在两文库表达量最高,且差异极显著。通路分析表明,ko00511、ko00052、ko01100、ko00030、ko00520 5个通路可能影响性激素的合成。测序获得安淮山羊垂体组织miRNA序列及表达谱,垂体组织miRNA表达丰富,且表达量各异。研究结果有助于理解miRNA在山羊垂体中调节性激素分泌的作用,并且所鉴别的miRNA有助于后续卵泡期~黄体期转变的相关研究。
miRNA;山羊;卵泡期;黄体期;垂体;Solexa
microRNA(miRNA)是一种内源性的18~22个核苷酸的非编码RNA,可以在转录后水平通过抑制mRNA翻译或促使miRNA降解进而调节基因的表达[1]。已有研究表明,在高等真核生物基因组中,已知miRNA约占1%,并且高达30%的蛋白编码基因受miRNA的调控[2]。miRNA的调控涉及到多种生理过程,包括细胞增殖[3-4]、细胞凋亡[5]、肿瘤发生[6]、生殖调控[7]、细胞分化[8]、新陈代谢[9]和激素的分泌[10]。在山羊的研究中,有关山羊乳腺miRNA的相关研究[11]、毛囊发育[12]和繁殖及肌肉方面的功能研究[13-14]较多。在垂体组织的研究已经证实EphA2是miR-26b的靶基因,并且成功建立miRNA的表达模式,为研究miR-26b在早期胚胎发育、垂体激素分泌和其他生殖功能中的研究提供基础[15]。在培养的垂体细胞中过表达miR-325-3p可抑制促黄体激素(LH)的分泌及其在细胞中的含量[16]。进一步的研究表明,促性腺激素释放激素(GnRH)通过miR-132/212和SIRT1-FOXO1通路促进促卵泡激素(FSH)的表达,这是第1个促性腺激素释放激素调节促性腺激素基因表达的microRNA通路[17]。在卵巢中部分miRNA的表达是由FSH调控的,并且FSH通过miRNA的网络调节卵泡的生成,垂体可通过分泌FSH调节卵泡的生成,从而调节山羊的繁殖[18]。
垂体是动物体最重要的内分泌腺,分泌多种激素,如生长激素、促性腺素、催产素、催乳素等。这些激素对代谢、生长、发育和生殖等有重要作用。对于垂体中参与调节生殖激素的miRNA的研究还较少。本研究通过高通量测序的方法,探测卵泡期和黄体期安淮山羊垂体中miRNA的表达和差异表达情况,预测其候选靶基因,并对差异表达miRNA的靶基因进行生物信息学分析。有助于理解miRNA在山羊垂体中调节性激素的分泌所起的作用,所鉴别的一些miRNA有助于后续卵泡期~黄体期转变的相关研究。
1 材料与方法
1.1 样品的采集
采集安淮山羊垂体组织样品6份,分成卵泡期组(FP)和黄体期组(LP),两组分别来自3只卵泡期的母羊垂体和3只黄体期的母羊垂体,6只山羊均为3岁半左右,已产过3胎。每组3只山羊垂体混合为一个池。在采集垂体组织样前,通过试情和B超观察山羊卵巢,符合条件的母羊屠宰后收集该羊的垂体组织样于液氮中暂时保存,后转移至-80 °C冰箱中长期冷冻保存。本试验中的6只安淮山羊均来自于合肥博大牧业科技开发有限责任公司。为排除试验中的其他因素的影响,各个试验羊只的体况和年龄等基本一致,在种羊场内统一采取该场的舍饲饲喂与管理制度。
1.2 RNA文库的构建
采用RNAiso Plus (TaKaRa)试剂盒提取山羊的垂体组织总RNA并进行质量检测。使用PAGE胶分离不同片段大小的RNA,切取18~30 nt的条带,回收Small RNA;配制5′接头连接体系,混匀离心,室温反应一定时间,再用PAGE 胶纯化回收5′连接产物;配制3′接头连接体系,混匀离心,室温反应一定时间,再用PAGE 胶纯化回收3′连接产物;配制反转录体系,在PCR 仪上室温反应一定时间,使连接产物反转录成双链,再配制PCR 反应体系,在PCR 仪上按照一定程序进行扩增。经过纯化和质检,达到要求后,基于Illumina Solexa测序平台的高通量测序,由深圳华大基因科技有限公司完成。
1.3 数据分析
小RNA测得raw data数据通过杂质的过滤和初步判断进行筛选。对过滤后的数据进行数据质量统计和长度统计。统计两样品间公共序列和特有序列的种类(用unique表示)及数量(用total表示)分布情况,并将所有sRNA与各类RNA,采用NCBI GenBank (ftp://ftp.ncbi.nlm.nih.gov/genbank/)和Rfam (http://rfam.janelia.org/)两个数据库进行比对注释[19]。通过Blast或Bowtie将sRNA和miRBase (http://www.mirbase.org/ftp.shtml)数据库比对,鉴定出已知miRNA用于后续分析。对未注释上任何RNA且比对上基因组外显子反义链、内含子、基因间区的sRNAs,通过选用软件Mirdeep筛选miRNA的生物特征预测出新miRNA。
对两个样品中表达的miRNA统计,分析样品间的表达量差异显著性,并分别使用log2-ratio、Scatter plot图比较两者共同表达的miRNA表达量的差异[20]。
利用cluster软件,依据显著差异表达基因差异倍数的log2值进行聚类。采用RNAhybrid软件对miRNA进行靶基因预测。通过GO功能显著性富集分析确定候选靶基因行使的主要生物学功能。并以KEGG中的通路为单位进行通路显著性富集分析,筛选与整个参考基因相比较在候选靶基因中显著性富集的通路[21]。FDR≤0.05的通路被认为在候选靶基因中显著富集。通过通路显著性富集分析能确定候选靶基因参与的最主要生化代谢途径和信号转导途径。
1.4 荧光定量PCR验证
为验证高通量测序结果的准确性,本试验随机选取了5个miRNA进行qRT-PCR的验证试验。用1 μg的总RNA按照反转录试剂盒SYBR®Prime ScriptTMmiRNA RT-PCR试剂盒(TaKaRa, Japan)的说明书步骤来获得cDNA。在37 ℃孵育1 h,95 ℃灭活5 min,之后加入80 μL 的RNase free H2O稀释反应体系至100 μL,并储存于-20 ℃冰箱中,以产生qRT-PCR的模板。qRT-PCR的特异性上游引物是根据各个所选miRNA的自身序列进行设计,其下游引物为通用引物,引物由生工生物工程(上海)股份有限公司合成(表1)。选择GAPDH作为内参基因,每个miRNA指标经qRT-PCR扩增后,得到各个指标的CT值,使用2-(CTmiRNA-CT5sRNA)法计算不同miRNA之间的相对表达量。
表1 荧光定量PCR扩增引物序列
Table 1 Primer sequences of real-time PCR

名称Name序列(5'-3')Sequence长度/ntLengthGC含量/%GCcontentchi-let-7fUGAGGUAGUAGAUUGUAUAGUU2231.8chi-miR-125aUCCCUGAGACCCUUUAACCUGU2250.0chi-miR-148aAAAGUUCUGAGACACUCCGACU2245.5chi-let-7aUGAGGUAGUAGGUUGUAUAGUU2236.4chi-miR-26aUUCAAGUAAUCCAGGAUAGGCU2240.9
2 结 果
2.1 测序数据的质量与分布
随机选择5条RNA-Seq显示在卵泡期垂体和黄体期垂体中存在显著性差异的miRNA,通过实时定量PCR对其检测结果进行验证。检测显示,5条差异miRNA,存在差异且差异趋势与RNA-Seq检测结果一致(图1)。
对过滤后的数据进行测序质量评估合格后,统计测序结果显示卵泡期山羊(FP)共得到11 319 069条原始序列,黄体期山羊(LP)共得到11 432 846条原始序列。经过去除杂质后,卵泡期文库得到11 162 888条纯净序列,占测得高质量序列的99.05%;黄体期文库得到11 016 438条纯净序列,占测得高质量序列的96.78%。这些纯净序列将用于后续的数据分析(表2)。
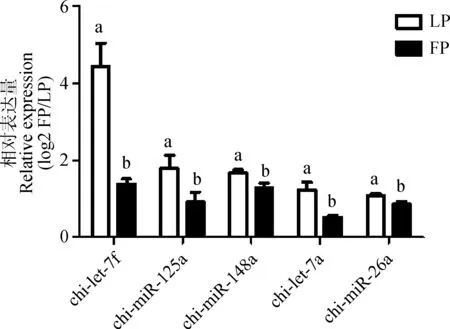
上标的字母表示在0.05显著水平上的比较结果Superscript letters indicate significant difference at the level of 0.05图1 5条差异miRNAs的RT-PCR结果Fig.1 The RT-PCR results of 5 differentially expressed miRNAs
表2 高通量测序文库的小RNA片段质量分类
Table 2 The classification of total small RNA tags by Solexa sequencing

分类Type卵泡期FP黄体期LP数量Count百分比/%Percent数量Count百分比/%PercentTotal_reads1131906911432846High_quality11269747100.0011382781100.003'adapter_null384580.34534190.47insert_null4700.003890.005'adapter_contaminants23670.0254350.05Smaller_than_18nt655200.583070842.70PolyA440.00160.00Clean_reads1116288899.051101643896.78
统计两个文库中序列的长度分布(图2),图2表明,卵泡期文库中序列长度分布主要位于20~30 nt,黄体期文库中序列长度分别主要位于20~23 nt;波峰与miRNA的典型长度22 nt一致,在卵泡期和黄体期文库中分别占26.42%和54.07%。动物体中成熟的miRNA序列是Dicer酶的酶切产物,长度分布是在22 nt左右,说明本试验的小RNA的测序文库长度分布基本囊括了各类miRNA。
通过统计两样品间公共序列和特有序列的种类及数量,可以得到两样品间公共序列和特有序列的分布情况(图3)。图3表明,两文库间的公共序列有20 504 157条占总序列的92.45%,特有序列种类分别有659 839条和149 547条,分别占有75.65%和17.15%。测序结果表明,两样品间序列种类的差异比较大,但公共部分的序列其表达是比较集中的,这说明两样品在测序整体上的一致性较好。

A.卵泡期; B.黄体期A.FP; B.LP图2 测序结果的序列长度分布Fig.2 Distribution of sequence lengths of the sequencing results

图3 公共(A)和特有(B)sRNA种数Fig.3 Number of total (A) and unique (B) sRNA tags
2.2 sRNA分类注释
将所有sRNA与各类RNA的比对情况进行总结,得到了卵泡期纯净序列共722 640种,黄体期纯净序列共212 348种。去除各类小RNA(rRNA、snRNA、snoRNA、tRNA)和重复序列后,参与后续的数据库比对的miRNA在卵泡期和黄体期中分别占36.44%和46.31%。但是在序列种类中,miRNA在卵泡期和黄体期文库中仅占0.37%和1.55%,其余大部分序列主要是被归类为未知序列。测序结果显示miRNA的表达量占主要部分,而种类最多的未知序列的表达量非常低(图4)。
2.3 已知miRNA比对和新miRNA预测
通过blast或bowtie将sRNA和miRBase 21.0数据库比对,由于数据库中已收录山羊miRNA的相关信息,本试验是将测序文库比对到山羊的miRNA的数据库中,鉴定出卵泡期和黄体期分别得到368种和371种已知miRNA,分别得到8 482 909和5 108 138的序列表达量。这些已知miRNA可用于后续分析。使用mirdeep(该软件适用动物)对未注释上任何RNA且比对上基因组外显子反义链、内含子、基因间区的sRNAs预测新miRNA,结果得到新miRNA卵泡期75种和黄体期125种,表达量分别为86 670和191 152。其中有46种在卵泡期和黄体期共同表达,且卵泡期和黄体期的新miRNA的表达量高于1 000拷贝数有5和8种,剩余大部分不高于100个拷贝数。
2.4 已知miRNA和新miRNA分析
2.4.1 已知miRNA和新miRNA的差异分析 对已知miRNA和新miRNA进行差异分析,为找出已知miRNA和新miRNA两种文库的差异性,首先对两种文库各自特异性表达miRNA进行分析。得到已知miRNA中,卵泡期和黄体期文库分别得到148和150种差异表达的miRNA,有147种miRNA为共同表达,卵泡期特异性表达1种,黄体期特异性表达3种;新miRNA中卵泡期文库和黄体期文库分别得到52和95种差异表达的miRNA,有27种miRNA两文库共同表达,卵泡期特异性表达的有25种,黄体期特异性表达的有68种。两种文库中大部分的miRNA表达量较低,而且在已知miRNA文库中特异性表达的miRNA不高于5。在新miRNA文库中,特异性表达的有novel-miR-176表达量达到1 388和novel-miR-189的表达量为1 416,剩余的大部分都不高于100。而表达量高且差异显著的miRNA中,各种miRNA的表达量差异较大,在两种文库中已知miRNA和新miRNA的表达量主要集中于少数几个miRNA中。已知miRNA中卵泡期和黄体期文库的表达量高于1 000的miRNA部分差异较大,且差异显著;新miRNA中卵泡期和黄体期文库分别有1个和3个miRNA的表达量高于1 000。见表3,4及图5。
表3 已知miRNA中表达量超过1 000且差异显著的miRNA
Table 3 Expressed level of higher than 1 000 and significant differently in the known miRNAs

miR_name卵泡期FPFP-expressedFP-stdmiR_name黄体期LPLP-expressedLP-stdchi-let-7f-5p11853420064.62chi-let-7f-5p1092904190313.30chi-let-7a-5p460537795.536chi-let-7a-5p26290845781.59chi-miR-10b-5p425217197.663chi-let-7e-5p16386728535.04chi-miR-129-3p143082421.96chi-miR-423-5p8474714757.45chi-let-7e-5p97321647.37chi-miR-222-3p252924404.23chi-miR-423-5p94161593.88chi-miR-296-3p189313296.56chi-miR-222-3p5244887.67chi-miR-200b139882435.81chi-miR-296-3p2768468.55chi-miR-10b-5p98091708.09chi-miR-200b3208543.03chi-miR-128-3p5447948.52chi-miR-485-5p3946687.14chi-miR-27a-3p3082536.69chi-miR-224-5p2690468.42chi-miR-877-5p2004348.97chi-miR-126-3p1752305.09chi-miR-129-3p1405244.66
表4 新miRNA中表达量超过1 000的miRNA
Table 4 Expressed level of higher than 1 000 and significant differently in the novel miRNAs

miR_name卵泡期FPFP-expressedFP-stdmiR_name黄体期LPLP-expressedLP-stdnovel_miR_20178281325.070novel_miR_159442337702.531novel_miR_15968661162.229novel_miR_201220133833.242novel_miR_1891416239.691novel_miR_5568981201.186novel_miR_1761388241.700

A、C.卵泡期文库中所有序列和特有序列分布;B、D.黄体期文库中所有序列和特有序列分布A, C.Total number of reads and unique sequences in the Fols;B,D.Total number of reads and unique sequences in the Luts图4 高通量测序中小RNA的分类组成Fig.4 Composition of small RNA classes of the sequencing

图5 已知miRNA (A)和新miRNA (B)表达的差异散点图Fig.5 Differences of miRNA expression in the known (A) and novel miRNA libraries (B)
2.4.2 已知miRNA和新miRNA的表达模式聚类分析 将两种文库差异表达miRNA进行聚类分析后,将表达模式相似的miRNA进行相互聚类,聚类图中绿色表示miRNA在黄体期文库中的表达水平高于卵泡期,红色表示miRNA在卵泡期文库中的表达水平高于黄体期,新miRNA文库中差异表达的miRNA聚类如图6。
2.4.3 已知miRNA和新miRNA的靶基因预测 对已知miRNA和新miRNA进行靶基因预测,使用RNAhybrid软件对已知miRNA进行靶基因预测。已知miRNA中卵泡期和黄体期文库差异表达的miRNA有151种,卵泡期和黄体期预测得到的靶基因位点数量为29 494;新miRNA中卵泡期文库和黄体期文库有120种差异表达的miRNA,卵泡期和黄体期预测得到的靶基因位点为29 494。

图6 新miRNA文库中差异表达miRNA聚类图Fig.6 Clustering of miRNAs differentially expressed in novel miRNA libraries
2.4.4 GO富集与KEGG分析 本试验GO富集情况分析中,差异表达的已知miRNA的GO富集分析结果显示,10 793个背景基因被映射到所处的细胞位置(Cellular component)部分,有2 045个差异表达的miRNA靶基因被映射到该部分,与背景基因的映射相比较,有2个条目(MHC protein complex,anchoring junction)显著富集(P-value<0.05);有10 487个背景基因被映射到分别描述基因的分子功能(Molecular function)部分,有1 955个miRNA的靶基因被映射到该部分,与背景基因的映射相比较,有1个条目(Receptor binding)显著富集;而有10 020个背景基因被映射到参与的生物过程(Biological process)部分,有1 909个miRNA的靶基因被映射到该部分,与背景基因的映射相比较,有2个显著富集条目(Membrane lipid metabolic process,hematopoietic progenitor cell differentiation)。
差异表达的新miRNA富集分析中各部分的背景基因与已知miRNA组相同。有2 435个miRNA的靶基因被映射到所处的细胞位置部分,有2个显著富集条目(Cell division site,cell division site part);有2 400个miRNA的靶基因被映射分别描述基因的分子功能部分,有2个条目显著富集(Ras GTPase binding,small GTPase binding);有2 285个miRNA的靶基因被映射到参与的生物过程部分,有3个条目显著富集(Ribonucleoprotein complex assembly,response to light stimulus,ribonucleoprotein complex subunit organization)。GO功能分析针对的是靶基因预测结果,通过对显著富集term中的miRNA进行GO功能分析,以基因的数量来推断miRNA的功能。结果发现差异表达的已知miRNA和新miRNA文库的功能分析结果图中Cell的百分比最多,说明样品中miRNA主要是靶向与cell相关的基因(图7)。
KEGG分析结果显示,已知miRNA组和新miRNA组中都有22 885个山羊背景基因,在已知miRNA和新miRNA文库中,分别被标注到283和294个生物学过程中。对于卵泡期和黄体期两文库所测数据中,已知miRNA和新miRNA文库中分别有4 251和5 241个miRNA的靶基因被标注到相关的生物学通路中;并且与背景基因所参与的生物学过程相比较,分别有26和2个显著富集通路(Q-value<0.05)。

图7 差异表达的新miRNA靶基因GO分析(LP-vs-FP)Fig.7 GO classification annotated for the target gene of novel miRNA (LP-vs-FP)
3 讨 论
山羊的经济效益主要取决于它的总生产力,母山羊的生产率更依赖于生殖力和产羔数[22-24]。但是,山羊的排卵率是比较低的,其遗传力系数主要介于0.09~0.14,山羊的排卵率很难通过常规育种方法改善排卵率低。因此,研究人员希望通过分子辅助育种技术与miRNA的研究来提高山羊的排卵率[25]。垂体可以通过分泌FSH、LH和GnRH等激素调节卵泡的分泌,从而调节山羊的卵泡期到黄体期的转变。通过对小鼠的下丘脑-垂体-卵巢轴研究发现,miR-200b和miR-429参与哺乳动物生殖的调节及调节排卵[26]。在猪的垂体细胞中,上调miR-361-3p表达抑制FSH的分泌,下调miR-361-3p的表达可促进FSH的分泌[27]。miR-21在卵巢颗粒细胞转变成黄体细胞的过程中具有抗细胞凋亡作用,敲低miR-21的表达会诱导颗粒细胞调亡,排卵率明显下降,这种作用依赖于LH的分泌[28]。测序发现卵泡期和黄体期文库,分别得到148和150种差异表达的miRNA,有147种miRNA共同表达。在卵泡期和黄体期两文库中,分别预测得到52和95种差异表达的miRNA,有27种miRNA两文库共同表达。垂体在动物生殖过程中起重要作用,本研究可进一步探索垂体中miRNA在性激素分泌中所起到的作用,为后续的研究奠定基础。
为寻找卵泡期和黄体期垂体中miRNA的差异表达情况,笔者建立了卵泡期和黄体期的miRNA的两个文库,用以寻找miRNA水平上的差异表达情况。发现各组特异性表达的miRNA参与相关通路的调节,但其表达量过低,而表达量高且差异显著的miRNA研究发现,已知miRNA中let-7f在两种文库中表达量都最高,且差异极显著;其次分别为let-7a、 let-7e,除let-7家族之外表达量较高的为miR-10b、miR-129、miR-423、miR-222。研究发现let-7家族的let-7a、let-7b、let-7c、let-7I在早期闭锁、逐步闭锁与健康猪卵泡相比卵巢的卵泡明显下降,而let-7G在卵泡闭锁呈高表达[29]。用猪垂体前叶细胞为模型,研究发现let-7a和let-7c可能起到调节FSH分泌的作用[27]。在前人对垂体的测序分析表明,miR-10b在上调基因中表达倍数最高,且在促性腺激素分泌垂体腺瘤中表达显著差异[30]。本次对于垂体的测序结果表明,let-7f的表达量在卵泡期文库显著高于黄体期,且在差异表达的已知miRNA中let-7家族在LP组中占总表达量的28.9%,在FP组中占总表达量的16.8%。说明let-7家族和miR-10b可能参与垂体性激素的分泌,为研究其在垂体性激素分泌的调节方面提供帮助。
GO富集情况分析表明,已知miRNA和新miRNA的GO富集分析分别有5和7个显著富集条目。在GO功能分析表明,结果发现差异表达的已知miRNA和新miRNA文库的功能分析结果表明Cell的百分比最多,说明样品中miRNA主要是靶向与cell相关的基因。KEGG分析结果显示,与背景基因所参与的生物学过程相比较,已知miRNA和新miRNA文库所有的miRNA靶基因参与的生物学过程,分别有26个和2个通路显著富集(Q-value<0.05)。研究发现IL-6/C/EBPβ,P53/P21和P16通路在D-半乳糖处理老化垂体细胞中会被激活,但是慢性雌激素的治疗这些通路可免疫垂体肿瘤[31]。D-半乳糖可导致大鼠亚急性衰老LH、FSH、GnRH明显上升[32]。富集通路中,ko00511:Other glycan degradation、ko00052:Galactose metabolism、ko01100:Metabolic pathways、ko00030:Pentose phosphate pathway、ko00520:Amino sugar and nucleotide sugar metabolism 5个通路可能会通过参与D-半乳糖的代谢从而影响性激素的合成。
4 结 论
对卵泡期和黄体期山羊的垂体进行小RNA文库的构建,分别得到148种和150种已知miRNA,有147种miRNA共同表达。在卵泡期和黄体期两文库中,分别预测得到59和95种新miRNA,有27种miRNA两文库共同表达。卵泡期和黄体期文库中,已知miRNA中,let-7f在两文库差异极显著,且表达量最高,其次分别为let-7a、miR-10b和let-7e;新miRNA中novel-miR-159在两文库差异极显著,且表达量最高,其次为novel-miR-55。通路分析表明,ko00511、ko00052、ko01100、ko00030、ko00520通路可能影响性激素的合成。
[1] BARTEL D P. microRNAs: genomics, biogenesis, mechanism, and function[J].Cell, 2004, 116(2):281-297.
[2] HU S J, REN G, LIU J L, et al. microRNA expression and regulation in mouse uterus during embryo implantation[J].JBiolChem, 2008, 283(34):23473-23484.
[3] HUANG W,LI J,GUO X,et al. miR-663a inhibits hepatocellular carcinoma cell proliferation and invasion by targeting HMGA2[J].BiomedPharmacother,2016, 81:431-438.
[4] WANG F, ZHANG H, XU N, et al. A novel hypoxia-induced miR-147a regulates cell proliferation through a positive feedback loop of stabilizing HIF-1α[J].CancerBiolTher, 2016, 17(8):790-798.
[5] DING G C,CHEN M,WANG Y X,et al.microRNA-128a-induced apoptosis in HTR-8/SVneo trophoblast cells contributes to pre-eclampsia[J].BiomedPharmacother, 2016, 81:63-70.
[6] WANG X, XIA Y.microRNA-328 inhibits cervical cancer cell proliferation and tumorigenesis by targeting TCF7L2[J].BiochemBiophysResCommun, 2016, 475(2):169-175.
[7] LING Y H, REN C H, GUO X F, et al. Identification and characterization of microRNAs in the ovaries of multiple and uniparous goats (Caprahircus) during follicular phase[J].BMCGenomics, 2014, 15:339.
[8] ANDREAS E,HOELKER M,NEUHOFF C,et al.microRNA 17-92 cluster regulates proliferation and differentiation of bovine granulosa cells by targeting PTEN and BMPR2 genes[J].CellTissueRes, 2016. 366(1):219-230.
[9] DAHLMANS D, HOUZELLE A, SCHRAUWEN P, et al. Mitochondrial dynamics, quality control and miRNA regulation in skeletal muscle: implications for obesity and related metabolic disease[J].ClinSci(Lond), 2016, 130(11):843-852.
[10] BUTTERWORTH M B. microRNAs and the regulation of aldosterone signaling in the kidney[J].AmJPhysiolCellPhysiol, 2015, 308(7):C521-C527.
[11] CHEN Z,LUO J,MA L,et al.miR130b-regulation of PPARγ coactivator- 1α suppresses fat metabolism in goat mammary epithelial cells[J].PLoSOne, 2015, 10(11):e0142809.
[12] 江 玮,范一星,乔 贤,等.皮肤毛囊发育的转录组研究进展[J].遗传,2015, 37(6):528-534.
JIANG W, FAN Y X, QIAO X, et al. The transcriptome research progresses of skin hair follicle development[J].Hereditas(Beijing), 2015, 37(6):528-534.(in Chinese)
[13] ZHANG X D, ZHANG Y H, LING Y H, et al. Characterization and differential expression of microRNAs in the ovaries of pregnant and non-pregnant goats (Caprahircus)[J].BMCGenomics, 2013, 14:157.
[14] YUAN B, HAN D X, DAI L S, et al. A comprehensive expression profile of micrornas in rat’s pituitary[J].IntJClinExpMed, 2015, 8(8):13289-13295.
[15] YUAN B, YU W Y, DAI L S, et al. Expression of microRNA-26b and identification of its target gene EphA2 in pituitary tissues in Yanbian cattle[J].MolMedRep, 2015, 12(4):5753-5761.
[16] NEMOTO T, MANO A, SHIBASAKI T. Increased expression of miR-325-3p by urocortin 2 and its involvement in stress-induced suppression of LH secretion in rat pituitary[J].AmJPhysiolEndocrinolMetab, 2012, 302(7):E781-E787.
[17] LANNES J, L’HTE D, GARREL G, et al. Rapid communication: A microRNA-132/212 pathway mediates GnRH activation of FSH expression[J].MolEndocrinol, 2015, 29(3):364-372.
[18] YAO N, LU C L, ZHAO J J, et al. A network of miRNAs expressed in the ovary are regulated by FSH[J].FrontBiosci(Landmark), 2009,14:3239-3245.
[19] HAO D C, YANG L, XIAO P G, et al. Identification of taxus microRNAs and their targets with high-throughput sequencing and degradome analysis[J].PhysiolPlant, 2012, 146(4):388-403.
[20] HUANG J, JU Z, LI Q, et al. Solexa sequencing of novel and differentially expressed microRNAs in testicular and ovarian tissues in Holstein cattle[J].IntJBiolSci, 2011, 7(7):1016-1026.
[21] KANEHISA M, ARAKI M, GOTO S, et al. KEGG for linking genomes to life and the environment[J].NucleicAcidsRes, 2008, 36(Database issue):D480-D484.
[22] MASCARENHAS R, SIMOES NUNES A, ROBALO SILVA J. Cyclic reproductive activity and efficiency of reproduction in Serrana goats[J].AnimReprodSci, 1995, 38(3):223-229.
[23] KHANUM S A, HUSSAIN M, KAUSAR R. Assessment of reproductive parameters in female Dwarf goat (Caprahircus) on the basis of progesterone profiles[J].AnimReprodSci, 2007, 102(3-4):267-275.
[24] ZHANG C Y, CHEN S L, LI X, et al. Genetic and phenotypic parameter estimates for reproduction traits in the Boer dam[J].LivestSci, 2009, 125(1):60-65.
[25] MCBRIDE D, CARRÉ W, SONTAKKE S D, et al. Identification of miRNAs associated with the follicular-luteal transition in the ruminant ovary[J].Reproduction, 2012, 144(2):221-233.
[26] HASUWA H, UEDA J, IKAWA M, et al. miR-200b and miR-429 function in mouse ovulation and are essential for female fertility[J].Science, 2013, 341(6141):71-73.
[27] YE R S, XI Q Y, QI Q, et al. Differentially expressed miRNAs after GnRH treatment and their potential roles in FSH regulation in porcine anterior pituitary cell[J].PLoSOne, 2013, 8(2):e57156.
[28] CARLETTI M Z, FIEDLER S D, CHRISTENSON L K. microRNA 21 blocks apoptosis in mouse periovulatory granulosa cells[J].BiolReprod, 2010, 83(2): 286-295.
[29] CAO R, WU W J, ZHOU X L, et al. Expression and preliminary functional profiling of the let-7 family during porcine ovary follicle atresia[J].MolCell, 2015, 38(4): 304-311.
[30] LIANG S, CHEN L, HUANG H, et al. The experimental study of miRNA in pituitary adenomas[J].TurkNeurosurg, 2013, 23(6):721-727.
[31] ZHANG T H, ZHAO B H, LI J, et al. Pituitary gene expression differs in D-galactose-induced cell senescence and steroid-induced prolactinomas[J].MolMedRep, 2015, 11: 3027-3032.
[32] 牛嗣云, 韩广明,刘红艳, 等. 松花粉对雄性衰老大鼠性腺轴分泌功能的影响[J]. 河北医学, 2011,17(7):872-874.
NIU S Y, HAN G M, LIU H Y, et al. The effect of pine pollen on male aging rats gonad axis secretory function[J].HebeiMedicine, 2011,17(7):872-874.(in Chinese)
(编辑 程金华)
Comparative Profiling of Differentially Expressed microRNAs between the Follicular and Luteal Phases Pituitary of Goats
LING Ying-hui1,2,ZHU Long1,2,SUI Meng-hua1,2,ZHENG Qi1,2,WU Hao1,2,ZHANG Yun-hai1,2,ZHANG Xiao-rong1,2,3*
(1.CollegeofAnimalScienceandTechnology,AnhuiAgriculturalUniversity,Hefei230036,China;2.LocalAnimalGeneticResourcesConservationandBiobreedingLaboratoryofAnhuiProvince,Hefei230036,China;3.EngineeringResearchCenterofReproductionandBreedinginSheepofAnhuiProvince,Hefei230036,China)
In this study, we used Solexa sequencing to screen and analyze the differentially expressed microRNAs (miRNAs) of Anhuai goat pituitary tissues in the follicular phase (Fols) and luteal phase (Luts), and explore the role of pituitary at post-transcriptional level of Fols to Luts transition occurred in goats. In total, 11 319 069 and 11 432 846 raw reads were obtained from the ovaries of Anhuai goats in Fols and Luts, respectively, after eliminating impurity, 11 162 888 and 11 016 138 clean reads. 147 known miRNAs were co-expressed in the two different periods phases, 148 and 150 known miRNAs were expressed in the ovary in the Fols and Luts, respectively. In addition, 27 novel miRNAs were co-expressed in the two phases, 52 and 95 novel miRNAs were expressed in the ovary in the Fols and Luts, respectively. Let-7f was the highest expressed significantly different known miRNAs in the two phases, and miR-159 was the highest expressed significantly different novel miRNAs in the two phases, which may participate in the follicular-luteal transition of Anhuai goats. In the KEGG pathway analysis, ko00511, ko00052, ko01100, ko00030 and ko00520 may be related to the synthesis of sex hormones. The study succeeded to construct the expression library of miRNAs which were abundant and differentially expressed in Anhuai goat pituitary tissues. The results will help to further understand the role of miRNAs which regulates secretion of hormone participate in goat pituitary. And some identified miRNAs will help to further understand the role of miRNAs in the regulation of follicular to luteal transition in goat ovaries.
miRNA;goat;follicular phase;luteal phase;pituitary;Solexa
10.11843/j.issn.0366-6964.2016.11.009
2016-06-30
国家自然科学基金项目(31301934;31372310);安徽省现代农业产业技术体系(2016-2020)
凌英会(1981-),男,安徽安庆人,副教授,博士,主要从事动物遗传育种与繁殖研究,E-mail:caaslyh@163.com
*通信作者:章孝荣,教授,博士生导师,主要从事动物生殖调控研究, E-mail:zhangxiaorong01@163.com
S827.2
A
0366-6964(2016)11-2218-10
