病毒感染激活炎症小体的分子机制
2016-12-13高泽乾朱学亮张志东窦永喜
高泽乾,朱学亮,张志东,窦永喜
(中国农业科学院兰州兽医研究所,家畜疫病病原生物学国家重点实验室,兰州 730046)
病毒感染激活炎症小体的分子机制
高泽乾,朱学亮,张志东,窦永喜*
(中国农业科学院兰州兽医研究所,家畜疫病病原生物学国家重点实验室,兰州 730046)
炎症小体是宿主细胞应对外界刺激、特殊病原或细胞损伤相关分子产生的一类多聚蛋白复合物,可以直接导致宿主细胞发生炎性坏死,即细胞焦亡。炎症小体复合物主要由炎症信号识别受体、凋亡相关点样蛋白(ASC)和含半胱氨酸的天冬氨酸水解酶1(caspase-1)组成。炎症信号识别受体识别刺激信号后,自身发生寡聚化,并募集ASC和caspase-1,活化的caspase-1切割促炎症因子前体(pro-IL)-1β和IL-18,产生成熟的促炎细胞因子IL-1β和IL-18。根据炎症信号识别受体的种类,炎症小体主要分为两类,即核苷酸结合寡聚化结构域样受体(NLR)炎症小体和黑色素瘤缺乏因子2样受体(ALR)炎症小体。宿主细胞可以识别病毒的不同结构,如离子通道蛋白、非结构蛋白和病毒核酸等,并产生相应的炎症小体,进而激活后续炎症和免疫相关反应导致细胞焦亡。作者从病毒结构的角度出发,阐述了宿主细胞是如何应对病毒不同结构的刺激并产生相应的炎症小体。
病毒;细胞焦亡;炎症小体;NLRP3;AIM2;IFI16
1 炎症小体概述
炎症小体的产生是宿主细胞应对外界病原入侵的重要手段。宿主细胞识别病原炎症刺激信号后,炎症小体进行组装并激活含半胱氨酸的天冬氨酸水解酶1(caspase-1)和促进促炎细胞因子白细胞介素(interleukin, IL)-1β和IL-18的成熟。促炎细胞因子促进炎症细胞因子产生并诱发一系列免疫反应,导致细胞发生炎性坏死,即细胞焦亡。细胞焦亡概念最早由A. Zychlinsky等于1992年提出,其本质上是一种细胞程序性死亡。焦亡早期细胞膜或细胞器膜等质膜上会形成小阳离子渗透空隙(即质膜孔),引起质膜两侧离子梯度消失,造成渗透溶胀和裂解,最终细胞释放内容物并造成强烈的炎症反应,还会引起多种自身免疫炎症和自身免疫病[1]。经典炎症小体的组装主要依赖两种炎症信号识别分子,即核苷酸结合寡聚结构域(nucleotide binding oligomerization domain, NOD)样受体和黑色素瘤缺乏因子2(absent in melanoma 2, AIM-2)样受体[2]。人类基因组编码22个NOD样受体(nucleotide-binding oligomerization domain-like receptor,NLR),但只有核苷酸结合寡聚化结构域样受体1 (NOD-like receptor family pyrin domain-containing 1, NLRP1)、NLRP3、NLRP6、NLRP7、NLRP12和NAIP/NLRC4被证实参与相应炎症小体组装[3-8]。其他NOD样受体的生物学功能仍不清楚。AIM2样受体主要包括AIM2和干扰素诱导蛋白16(interferon inducible protein 16, IFI16),均可组装成炎症小体,尤其是在宿主细胞识别病毒核酸炎症信号过程中发挥作用[9-10]。
NLRP3炎症小体是目前研究最为透彻的炎症小体。NLRP3属于NLR家族,含有多个功能结构域,包括热蛋白结构域(pyrin domain,PYD)、杆状病毒抑制因子细胞凋亡重复域(baculovirus inhibitor of apoptosis repeat domain,BIR)、中央核苷酸结合寡聚化结构域(central nucleotide-binding and oligomerization domain,NACHT)和富亮氨酸重复序列(leucine-rich repeats,LRRs)(图1)。NLRP3通过其N端的PYD和其他含PYD形成异源多聚体,从而介导信号传输。NLRP3分子之间通过NACHT结构域发生寡聚化,组成炎症小体的支持结构,该过程是炎症小体形成的关键步骤。其C端的LRRs结构域负责识别病原结构成分。NLRP3接收到刺激信号后通过NACHT结构域形成同源多聚体,然后其PYD结构域直接和凋亡相关点样蛋白(apoptosis associated speck like protein containing a C-terminal caspase recruitment domain,ASC)的PYD结构域结合。然后通过ASC的 C端半胱天冬酶募集域(C-terminal caspase recruitment domain,CARD)与caspase-1的CARD结构域结合形成异源二聚体并激活caspase-1,活化的caspase-1切割促炎因子前体pro-IL-1β和pro-IL-18转变为成熟的IL-1β和IL-18(图2)[11]。由于NLRP3缺少CARD,所以通过ASC蛋白和caspase-1间接发生作用。NLR家族中NLRP1也参与组装炎症小体形成NLRP1炎症小体。由于NLRP1 N端含有CARD结构域,因此NLRP1能以非依赖ASC方式激活caspase-1,促进促炎因子IL-1β和IL-18前体的分泌和成熟[12]。

图1 炎症小体复合体各蛋白质结构模式Fig.1 Domain organization of inflammasome component proteins
AIM2结构较为简单,属于黑色素瘤缺乏因子2样受体(absent in melanoma 2-like receptor, ALR)家族,只有PYD和HIN-200两个结构域。AIM2 C端HIM-200结构域识别dsDNA(double stranded DNA)行使信号识别功能,其N端PYD结构域可以和ASC蛋白的PYD直接结合。然后ASC的CARD与caspease-1的CARD结合,组成AIM2炎症小体,促进促炎因子前体pro-IL-1β和pro-IL-18转变为成熟形式[13]。IFI16是ALR家族的另一成员,具有1个PYD结构域和2个HIN-200结构域,IFI16的HIN-200结构域可以识别病毒的DNA和RNA结构,促进IFI16炎症小体的组装。和AIM2炎症小体、NLRP3炎症小体不同,IFI16氨基酸序列中还有核定位信号,所以IFI16炎症小体既可在细胞质中完成组装,又可在细胞核内完成组装[10]。
病毒入侵宿主细胞后,病毒的多种成分被相应的炎症信号识别受体所识别,信号经多种接头分子传输至炎症小体,炎症小体完成组装后诱导促炎因子前体的分泌和成熟。目前已通过实验证实,能够激活炎症小体组装的病毒成分包括病毒核酸、病毒编码离子通道蛋白和一些非结构蛋白。笔者将对病毒激活炎症小体组装的分子机制进行综述,为病毒激活炎症小体相关研究奠定基础,并为相关药靶筛选和新药物开发提供新思路。
2 炎症小体激活机制
炎症小体的激活模型主要有三种。第一种,离子通道模型。离子通道模型主要分为钾离子通道模型和钙离子通道模型。钾离子模型主要表现为,当细胞膜外ATP浓度上升时,可以通过P2X7 ATP依赖性离子通道和膜的泛连接蛋白1(pannexin-1)孔隙促进钾离子外流,这样会促进病原相关分子模式(pathogen associated molecular patterns, PAMPs)和损伤相关分子模式(damage associated molecular patterns, DAMPs)进入细胞内,进而促进NLRP3炎症小体的组装和激活[14]。钙离子模型表现为,当细胞膜外Ca2+浓度升高时,磷脂酰肌醇/Ca2+信号通路被激活,钙敏感受体(calcium sensing receptors, CaSRs),G蛋白偶联受体C家族6A(G protein coupled receptor family C, group6, subtype A, GPRC6A)和磷脂酶C(phospholipase C,PLC)等活化后会分解磷脂酰肌醇-4,5-二磷酸(phosphatidylinositol-4,5-bisphosphate,PIP2)产生二脂酰甘油(diacylglycerol,DAG)和三磷酸肌醇(inositol triphosphate,InsP3)。InsP3通过结合其在内质网(endoplasmic reticulum,ER)上的受体,促进ER中贮存的Ca2+进入细胞质,进而促进炎症小体的组装和IL-1β的产生[15-17]。激活的CaSRs也可以通过降低细胞内环化腺苷酸(cyclic adenosine 3′,5′-monophosphate, cAMP)促进NLRP3炎症小体的激活[16]。第二种,线粒体模型。NLRP3炎症小体的激活因子可以通过激活NADPH氧化酶诱导线粒体产生活性氧(reactive oxygen species,ROS),而ROS与成熟促炎因子IL-1β的外排有紧密联系[18]。将与ROS产生直接相关的电压依赖性阴离子通道(voltage-dependent anion channel,VDAC)基因沉默后,发现炎症小体的组装激活受到抑制。细胞质中ROS的积累同样能促进钾离子的外流从而进一步激活NLRP炎症小体[19]。所以线粒体产生的ROS并不是直接在炎症小体组装和激活中发挥作用。同时,线粒体上脂质双磷脂酰甘油也参与调控炎症小体组装和激活过程,其直接结合NLRP3形成复合物,抑制NLRP3炎症小体的组装[20]。第三种,溶酶体破裂模型。NLRP3炎症小体激活因子(如二氧化硅、石棉等)通过吞噬作用进入细胞内,会诱导溶酶体破裂和内容物的释放,尤其是组织蛋白酶B(cathepsin B),在NLRP3炎症小体激活过程中起着重要作用[21]。
3 病毒结构成分激活炎症小体的机制
病毒侵入宿主细胞后,将宿主细胞的核酸和蛋白质合成等系统为自身所用,产生新的病毒粒子。病毒复制过程中的产生的多种结构成分可以激活炎症小体的组装,引起宿主细胞强烈的免疫反应,造成宿主细胞焦亡。这些引起炎症小体激活的病毒组分主要分为病毒粒子通道蛋白(Viroporin)、病毒核酸和病毒非结构蛋白。
3.1 病毒编码Viroporin激活炎症小体
Viroporin是病毒编码的一种小分子,疏水,多聚化后形成孔状结构的病毒蛋白质,在病毒入侵、复制、组装和释放等重要过程发挥重要作用。Viroporin基因编码50~120氨基酸,通常包括1到2个跨膜结构域(trans-membrane domain,TM),Viroporin穿透磷脂分子层后可以形成离子通道,暴露在细胞膜外的区域与相应的病毒或宿主蛋白质发生作用,从而改变细胞膜的通透性[22-23]。
根据TM的数量,Viroporin蛋白可以分为I和II家族。每个家族根据其N端和C端的膜穿透特性和拓扑结构又分为两个亚家族。I家族含有1个 TM结构域,II家族含有2个TM结构域。I家族中Viroporin蛋白N端分布在细胞质内为A亚家族,而N端位于细胞外的为B亚家族。 II型Viroporin蛋白中C端和N端均位于细胞质中为A亚家族,均位于细胞膜外为B亚家族。此外还有研究报道存在III型Viroporin家族,该家族还有3个TM结构域,其生化特性有待研究。由于该结构特点,Viroporin很容易发生多聚化[22]。一旦Viroporin蛋白在细胞膜表面发生多聚化,就会和其他蛋白质共同组成离子通道蛋白复合体。通道蛋白复合体的形成是病毒入侵和释放过程中非常重要的阶段。
正黏病毒科A型流感病毒侵染细胞后,宿主细胞通过TLR7识别病毒基因组RNA,促进IL-1β和IL-18基因转录和前体的合成[24]。流感病毒编码的M2蛋白属于I型Viroporin,是一种质子特异性离子通道,在病毒入侵和新病毒粒子合成过程中扮演重要角色。M2蛋白主要定植于高尔基体上,可以将H+转运出细胞器内腔,造成高尔基体和细胞质的H+浓度失衡,这与NLRP3炎症小体激活直接相关。同时,将M2蛋白第37位氨基酸由组氨酸突变为甘氨酸后,M2蛋白获得了转运Na+和K+能力,进一步增强了细胞中炎症小体激活的程度,这表明不仅是H+,其他离子如Na+和K+等离子浓度失衡均能引起NLRP3炎症小体激活[25]。
微RNA病毒科成员脑心肌炎病毒(encephalomyocarditis virus,EMCV)侵入鼠树突状细胞和巨噬细胞后,能够激活NLRP3炎症小体的组装。单独将其以单链正义RNA形式的基因组转染细胞后可以引起巨噬细胞I型干扰素的分泌,但检测不到表达量呈显著上升的IL-1β和IL-18。EMCV非结构蛋白2B属于II型Viroporin,被证实定植于细胞内质网和高尔基体膜上,在鼠骨髓源巨噬细胞中进行过表达,可以使细胞器中的钙离子浓度降低,促使NLRP3炎症小体组装和IL-1β的分泌。2B蛋白的过表达不能通过线粒体ROS和溶酶体组织蛋白酶B途径引起NLRP3炎症小体组装和IL-1β分泌。除EMCV外,该属其他病毒(如脊髓灰质炎病毒和肠病毒71)编码的2B蛋白均可以引起NLRP3炎症小体的组装[26]。
Viroporin除了可以造成离子浓度失衡外,还有研究推测其可能通过线粒体活性氧ROS产生来激活炎症小体组装,但具体机制还不清楚,需要进一步探索[27-28]。
3.2 病毒核酸激活炎症小体的分子机制
病毒入侵宿主细胞后,其基因组释放到细胞质中,病毒基因组会被相应的炎症信号识别受体识别,激活相应炎症小体的组装。
A型流感病毒除编码Viroporin外,其基因组RNA同样可以激活NLRP3炎症小体。将病毒RNA类似物poly(I:C)接种于野生鼠鼻腔内可引起温和的炎症反应,对比将poly(I:C)接种于NLRP3缺陷型小鼠鼻腔,IL-1β呈显著下调趋势。采用shRNA方法发现PYCARD和NLRP3蛋白对于poly(I:C)诱导IL-1β产生是必需的。用富含GU的单链RNA(ssRNA40)刺激细胞同样可以引起NLRP3和PYCARD依赖性的IL-1β分泌。这表明细胞可以针对RNA病毒基因组产生炎症反应。通过采用药物拮抗剂阻断特殊信号通路的方法,发现DAMP和组织蛋白酶B与NLRP3介导的细胞死亡和IL-1β分泌直接相关。采用同样的方法发现线粒体ROS的产生也是炎症小体激活的必需步骤,但病毒核酸激活NLRP3炎症小体的具体机制仍不清楚[29]。流感病毒RNA可以激活NLRP3炎症小体组装,但NLRP3炎症小体的任何组分都不能直接结合病毒核酸,因此存在相应的核酸信号识别分子协助诱导NLRP3炎症小体的组装,如DDX19A可以直接结合动脉炎病毒RNA,协助激活NLRP3炎症小体的组装[30]。将丙型肝炎病毒(hepatitis C virus,HCV)的RNA基因组polyU/UC区段刺激THP1细胞,同样可以促进IL-1β mRNA的表达,但其具体的炎症小体激活机制仍不清楚[31-32]。除RNA病毒基因组可以激活炎症小体外,DNA病毒基因组也可以激活炎症小体。如痘苗病毒等正痘病毒属病毒侵入宿主细胞后,其基因组dsDNA可被细胞质中的炎症信号识别受体AIM2识别,促进AIM2炎症小体组装与IL-18和IL-1β促炎细胞因子的产生(图2)[33]。同样,细胞核中的病毒核酸也可以被相应的炎症信号识别受体识别,在细胞核中组装和激活相应的炎症小体。如,ALR家族的IFI16不仅在细胞质中识别单纯性疱疹病毒(herpes simplex virus,HSV)-1和卡波西肉瘤相关疱疹病毒(Kaposi′s sarcoma-associated herpesvirus, KSHV)的基因组DNA,同时也在细胞核中识别其基因组DNA,激活IFI16炎症小体[34-35]。病毒核酸是通过哪些接头蛋白将刺激信号传递至相应炎症信号识别受体的机制仍然不清楚,仍需进一步探索。
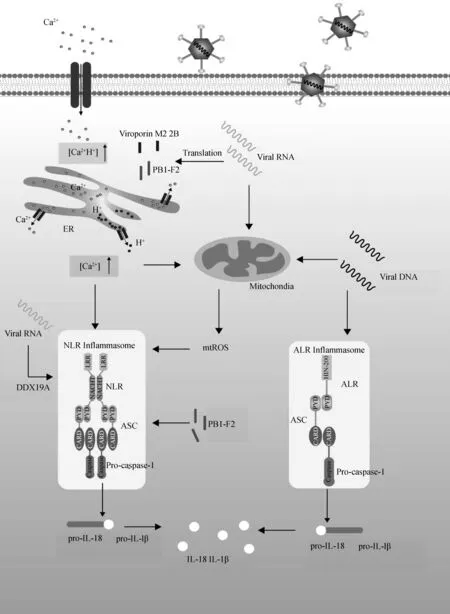
图2 病毒各组分激活炎症小体示意Fig.2 Inflammasome activation by diverse viral components
3.3 病毒非结构蛋白激活炎症小体
病毒编码的非结构蛋白也可以激活炎症小体。有研究发现,A型流感病毒编码的PB1-F2蛋白可以激活NLRP3炎症小体。该研究证实PB1-F2蛋白通过溶酶体破裂模型激活NLRP3炎症小体,诱导成熟IL-1β的产生[36]。病毒非结构蛋白激活炎症小体的研究非常少,有待进一步探索。
4 病毒结构抑制炎症小体激活的分子机制
病毒入侵宿主细胞后,宿主细胞为应对病毒感染,在持续的进化压力下,产生了多种炎症信号识别受体,它们能够识别病毒的不同结构,获得识别信号后激活相应的炎症小体,产生促炎症细胞因子,诱发细胞焦亡,从而抑制病毒繁殖和扩散。但是,病毒同样进化出相应的防护机制,主要途径是通过其非结构蛋白来抑制炎症小体的激活。病毒编码的多种非结构蛋白直接抑制炎症小体激活途径中的关键蛋白质,造成抑制效应。
病毒非结构蛋白可以和炎症小体激活途径关键蛋白质直接结合来抑制炎症小体的产生。如人巨细胞病毒(human cytomegalovirus,HCMV)编码的pUL83蛋白可以通过自身的PAD结构域直接和IFI16 PYD结构域结合,阻止IFI16多聚化,进而抑制IFI16炎症小体的产生[37];痘苗病毒编码的F1L蛋白也会直接结合NLRP1抑制其功能;麻疹病毒V蛋白可以直接抑制NLRP3蛋白,抑制IL-1β的产生[38]。病毒非结构蛋白通过泛素化降解来抑制炎症小体的激活。如,HSV-1编码一种E3泛素化连接酶ICP0,该蛋白质可以直接靶向IFI16,造成IFI16的泛素化降解[39]。由于炎症小体的炎症信号识别受体在细胞中的定位可通过乙酰化或磷酸化决定,所以病毒的非结构蛋白也可以通过对激活途径中的关键蛋白质进行乙酰化或磷酸化来改变信号识别受体的细胞定位,造成炎症小体抑制。例如,HCMC编码的激酶pUL97可以磷酸化IFI16,造成IFI16由细胞核内转运至细胞质,因此IFI16不能识别细胞核中HCMC的病毒DNA,无法激活IFI16炎症小体[40]。
5 总结与展望
病毒感染宿主细胞和炎症小体的组装激活是一个极其复杂的生物学过程。宿主细胞通过识别病毒的不同结构成分,激活相应的炎症小体和免疫反应,造成细胞焦亡,从而抑制病毒的繁殖。同时,病毒可以利用自身非结构蛋白来抑制炎症小体的组装和激活,以逃避宿主细胞的防御机制。病毒炎症小体的激活途径非常复杂,且各激活模型之间并不是独立发挥激活炎症小体作用,而是相互联系和相互影响。例如,病毒感染过中,线粒体模型中ROS的积累可以促进NLRP3炎症小体的激活,也可以通过离子通道模型促进钾离子外流的方式进一步激活NLRP3炎症小体[41]。但各个激活途径之间相互联系的研究仍处于起步阶段,需深入研究。同时,病毒炎症信号识别受体和相应接头分子的鉴定是炎症小体研究中非常重要的环节。这对于理解不同病毒炎症信号激活相应炎症小体以及各个炎症小体激活之间的相互联系非常重要。如人类NLR家族有22个成员,小鼠NLR家族有34个成员,大部分成员在炎症小体激活过程中的功能是未知的,需要进一步探索。病毒激活炎症小体的研究加深了我们对于宿主细胞抵抗外界病原的理解,为疾病治疗和药物开发提供新的思路。
[1] LAROCK C N, COOKSON B T. Burning down the house: cellular actions during pyroptosis[J].PLoSPathog, 2013, 9(12):e1003793.
[2] CHEN I Y, ICHINOHE T. Response of host inflammasomes to viral infection[J].TrendsMicrobiol, 2015, 23(1):55-63.
[4] JO E K, KIM J K, SHIN D M, et al. Molecular mechanisms regulating NLRP3 inflammasome activation[J].CellMolImmunol, 2016, 13(2):148-159.
[5] WLODARSKA M, THAISS C A, NOWARSKI R, et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion[J].Cell, 2014, 156(5):1045-1059.
[6] KHARE S, DORFLEUTNER A, BRYAN N B, et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages[J].Immunity, 2012, 36(3):464-476.
[7] VLADIMER G I, WENG D, PAQUETTE S W, et al. The NLRP12 inflammasome recognizesYersiniapestis[J].Immunity, 2012, 37(1):96-107.
[8] VANCE R E. The NAIP/NLRC4 inflammasomes[J].CurrOpinImmunol, 2015, 32: 84-89.
[9] HORNUNG V, ABLASSER A, CHARREL-DENNIS M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC[J].Nature, 2009, 458(7237):514-518.
[10] XIAO T S. The nucleic acid-sensing inflammasomes[J].ImmunolRev, 2015, 265(1):103-111.
[11] LATZ E, XIAO T S, STUTZ A. Activation and regulation of the inflammasomes[J].NatRevImmunol, 2013, 13(6):397-411.
[12] MASTERS S L, GERLIC M, METCALF D, et al. NLRP1 inflammasome activation induces pyroptosis of hematopoietic progenitor cells[J].Immunity, 2012, 37(6):1009-1023.
[13] MAN S M, KARKI R, KANNEGANTI T D. AIM2 inflammasome in infection, cancer and autoimmunity: role in DNA sensing, inflammation and innate immunity[J].EurJImmunol, 2016, 46(2):269-280.
[14] KANNEGANTI T D, LAMKANFI M, KIM Y G, et al. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling[J].Immunity, 2007, 26(4):433-443.
[15] ROSSOL M, PIERER M, RAULIEN N, et al. Extracellular Ca2+is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors[J].NatCommun, 2012, 3:1329.
[16] LEE G S, SUBRAMANIAN N, KIM A I, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+and cAMP[J].Nature, 2012, 492(7427):123-127.
[17] MURAKAMI T, OCKINGER J, YU J, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome[J].ProcNatlAcadSciUSA, 2012, 109(28):11282-11287.
[18] CRUZ C M, RINNA A, FORMAN H J, et al. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages[J].JBiolChem, 2007, 282(5):2871-2879.
[19] ZHOU R, YAZDI A S, MENU P, et al. A role for mitochondria in NLRP3 inflammasome activation[J].Nature, 2011, 469(7329):221-225.
[20] IYER S S, HE Q, JANCZY J R, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation[J].Immunity, 2013, 39(2):311-323.
[21] HORNUNG V, BAUERNFEIND F, HALLE A, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization[J].NatImmunol, 2008, 9(8):847-856.
[22] SZE C W, TAN Y J. Viral membrane channels: Role and function in the virus life cycle[J].Viruses, 2015, 7(6):3261-3284.
[23] SCOTT C, GRIFFIN S. Viroporins: structure, function and potential as antiviral targets[J].JGenVirol, 2015, 96(8):2000-2027.
[24] MARTINON F, TSCHOPP J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases[J].Cell, 2004, 117(5):561-574.
[25] ICHINOHE T, PANG I K, IWASAKI A. Influenza virus activates inflammasomes via its intracellular M2 ion channel[J].NatImmunol, 2010, 11(5):404-410.
[26] ITO M, YANAGI Y, ICHINOHE T. Encephalomyocarditis virus viroporin 2B activates NLRP3 inflammasome[J].PLoSPathog, 2012, 8(8):e1002857.
[27] TRIANTAFILOU K, KAR S, VAKAKIS E, et al. Human respiratory syncytial virus viroporin SH: a viral recognition pathway used by the host to signal inflammasome activation[J].Thorax, 2013, 68(1):66-75.
[28] SEGOVIA J, SABBAH A, MGBEMENA V, et al. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection[J].PLoSOne, 2012, 7(1):e29695.
[29] ALLEN I C, SCULL M A, MOORE C B, et al. The NLRP3 inflammasome mediatesinvivoinnate immunity to influenza a virus through recognition of viral RNA[J].Immunity, 2009, 30(4):556-565.
[30] LI J, HU L, LIU Y, et al. DDX19A senses viral RNA and mediates NLRP3-dependent inflammasome activation[J].JImmunol, 2015, 195(12):5732-5749.
[31] BURDETTE D, HASKETT A, PRESSER L, et al. Hepatitis C virus activates interleukin-1beta via caspase-1-inflammasome complex[J].JGenVirol, 2012, 93(Pt 2):235-246.
[32] NEGASH A A, RAMOS H J, CROCHET N, et al. IL-1beta production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease[J].PLoSPathog, 2013, 9(4):e1003330.
[33] RATHINAM V A, JIANG Z, WAGGONER S N, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses[J].NatImmunol, 2010,11(5):395-402.
[34] KERUR N, VEETTIL M V, SHARMA-WALIA N, et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection[J].CellHostMicrobe, 2011, 9(5):363-375.
[35] JOHNSON K E, CHIKOTI L, CHANDRAN B. Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes[J].JVirol, 2013, 87(9):5005-5018.
[36] THOMAS P G, DASH P, ALDRIDGE J R Jr, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1[J].Immunity, 2009, 30(4):566-575.
[37] LI T, CHEN J, CRISTEA I M. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion[J].CellHostMicrobe, 2013, 14(5):591-599.
[38] GERLIC M, FAUSTIN B, POSTIGO A, et al. Vaccinia virus F1L protein promotes virulence by inhibiting inflammasome activation[J].ProcNatlAcadSciUSA, 2013, 110(19):7808-7813.
[39] ORZALLI M H, DELUCA N A, KNIPE D M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein[J].ProcNatlAcadSciUSA, 2012,109(44):E3008-17.
[40] DELL′OSTE V, GATTI D, GUGLIESI F, et al. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage ofinvitrohuman cytomegalovirus infection and is entrapped in the egressing virions during the late stage[J].JVirol, 2014, 88(12):6970-6982.
[41] ZHOU R, TARDIVEL A, THORENS B, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation[J].Natimmunol, 2010, 11(2):136-140.
(编辑 白永平)
The Mechanisms of Inflammasomes Activation by Viral Components
GAO Ze-qian, ZHU Xue-liang, ZHANG Zhi-dong, DOU Yong-xi*
(ViralDiseasesinGrazingAnimalsProgramme,StateKeyLaboratoryofVeterinaryEtiologicalBiology,LanzhouVeterinaryResearchInstitute,ChineseAcademyofAgriculturalSciences,Lanzhou730046,China)
Inflammasomes are multiprotein complexes that induce downstream immune responses to environmental stimuli, specific pathogens and host cell damage, leading to the pyroptotic cell death of host cells. Inflammasomes mainly consist of recognition receptors of inflammatory signals ASC (the adapter apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain) and pro-caspase-1. Once activated, inflammasomes can induce the activation of caspase-1 and maturation of inflammatory cytokines, including IL-1β and IL-18. Inflammasomes can be classed into the NLR (nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3) inflammasome and the ALR (absent in melanoma 2 like receptor) inflammasome on the basis of the types of inflammatory signals recognition receptors. Host cells can recognize different viral components, including viroporins, non-structural proteins, viral double-stranded DNA and viral single stranded RNA, by different inflammatory signals recognition receptors and promote the formation of responding inflammasomes. Herein, we outlined the mechanisms of inflammasome formation activated by diverse viral components, and therefore provide a reference for future research and development of novel antiviral drugs.
viruses; pyroptosis; inflammasomes; NLRP3; AIM2; IFI16
10.11843/j.issn.0366-6964.2016.11.003
2016-06-13
中国农业科学院创新工程基金;公益性行业(农业)科研专项(201303059)
高泽乾(1988-),男,河北邯郸人,硕士,主要从事分子生物学与免疫学的研究,E-mail:zeqian_gao@sina.cn
*通信作者:窦永喜,副研究员,硕士生导师,博士,E-mail:douyongxi@caas.cn
S852.4
A
0366-6964(2016)11-2167-08
