Co-Y共掺杂ZnO光电性质的第一性原理计算
2016-12-05范涛健袁俊辉杨永勇余念念王嘉赋
范涛健 袁俊辉 杨永勇 余念念 王嘉赋
(武汉理工大学理学院物理系,武汉430070)
Co-Y共掺杂ZnO光电性质的第一性原理计算
范涛健袁俊辉杨永勇余念念*王嘉赋*
(武汉理工大学理学院物理系,武汉430070)
采用基于密度泛函理论(DFT)的第一性原理平面波赝势法研究了本征ZnO、Co和Y单掺杂ZnO、Co-Y不同配位共掺杂ZnO的电子结构和光学性质。计算结果表明,在本文的掺杂浓度下,Co和Y单掺杂可以提高ZnO的载流子浓度,从而改善ZnO的导电性,Co-Y共掺时ZnO半导体进入简并状态,呈现金属性。Co掺杂ZnO会在可见光和近紫外区域发生吸收增强现象,而Y掺杂ZnO可以提高体系在紫外区域的吸收,其中由于Co离子和Y离子之间的协同效应,Co-Y共掺ZnO时体系对可见光和近紫外区域的光子能量吸收大幅增加,因此Co-Y共掺杂ZnO可以用于制作光电感应器件。
ZnO,第一性原理,电子结构,光学性质
0 引言
作为一种新型的宽禁带(Eg=3.37 eV)化合物半导体材料,ZnO具有大光电耦合率、低介电常量,优良的压电、光电特性以及高的化学稳定性,同时它还具有高达60meV的激子结合能,是在紫外和蓝光发射方面很有前途的新型光电子材料[1-3]。目前很多研究都主要是通过对ZnO的元素掺杂使其光电改性进而投入实际应用。例如,ZnO中添加Mg,Ga,Al等元素可以实现对ZnO电阻系数的调控,改善其导电性能[4-6]。在ZnO中掺入Fe,Mn,Ni等过渡金属原子就能形成稀磁半导体材料[7-9]。郭少强等[10]的研究表明V掺杂会使ZnO体系的最小光学带隙增宽,同时吸收带边向低能级方向移动。刘玮洁等[11]研究了不同价态稀土元素Yb掺杂ZnO的光学性质,发现掺杂体系的吸收带边发生明显红移并且Yb2+的吸收效果更好。同时Co、Cu、Y作为普遍的光学掺杂元素,其掺杂ZnO后对体系的光学性质有明显的改善。包秀丽等[12]发现Co离子的引入会导致ZnO吸收谱中出现新的吸收峰,有蓝移现象发生。文献[13]采用溶胶-凝胶旋涂法制备了Co掺杂ZnO薄膜,观测了不同浓度Co样品的PL谱,发现其中出现了较弱的绿光发射和较强的蓝光双峰发射。曲盛薇等[14]在理论与实验上对Cu掺杂ZnO做了研究,发现Cu掺杂显著改变了ZnO薄膜在近紫外及可见光波段的吸收特性,其光学带隙随着Cu掺杂量的增加有所减小。Heo等[15]通过实验发现Y掺杂ZnO是n型掺杂,掺杂后会减小体系的电阻率,增强导电性;Zheng等[16]发现在与纯氧化锌纳米颗粒相比适量浓度的Y掺杂氧化锌在紫外区域的发射谱会有极大的增强。另外,在Cu-Co共掺ZnO与Cu-Y共掺ZnO体系中,也会由于协同效应而表现出吸引人的性质。吴定才等[17]用凝胶法制备了Cu-Co共掺杂的ZnO薄膜,发现Cu、Co掺杂会通过对ZnO薄膜中锌空位、锌填隙和氧空位等缺陷的影响实现对样品蓝光-绿光发光带强度的调节。何静芳等[18]则通过计算发现Cu-Co共掺杂ZnO对太阳光的吸收大幅增加是来源于Cu和Co离子间的协同效应。Anandan等[19]采用溶胶-凝胶方法制备了不同浓度的Y-Cu共掺杂ZnO材料并研究了其光学吸收,发现共掺体系对可见光和近紫外区域的光子能量吸收大幅增加,在光电感应器件的制作方面具有广阔的应用前景;而袁俊辉等[3]则基于第一性原理计算从理论上研究了Y-Cu共掺杂ZnO后体系光学性质改变的本质,为今后的实验提供了理论支撑。Co和Cu的光学性质有相似性,而且Co3+与Cu2+的离子半径接近(0.72 nm,0.73 nm),因此研究Y-Co不同配位共掺杂ZnO的电子结构和光学性质对于寻找新型光学材料具有重要意义。
本文采用基于密度泛函理论的第一性原理平面波赝势法,研究了Co和Y共掺杂ZnO的电子结构和光学性质,并与本征ZnO、Co掺杂ZnO和Y掺杂ZnO的结果进行了对比分析,希望可为制备高掺杂特性ZnO提供一定的理论支撑。
1 计算方法
ZnO有两种晶体结构,本文采用更为稳定的六方纤锌矿结构,空间群为P63mc,其晶胞由O的六角密堆积和Zn的六角密堆积在c轴方向反嵌套而成,ZnO晶格常数的实验值a=b=0.324 9 nm,c=0.520 6 nm,c/a=1.602。计算中我们建立了包含32个原子的2×2×2的ZnO超胞。晶胞掺杂方式是Y、Co原子替换ZnO超胞中的Zn原子。文献[12]中研究了Co单掺杂浓度分别为6.25%、12.5%、25%的掺杂ZnO结构。袁俊辉等[3]研究了掺杂浓度为6.25%的Y单掺杂ZnO。本文采用单掺杂时形成浓度为6.25%(原子分数)的掺杂ZnO结构,在计算之前首先对不同元素掺杂的ZnO进行几何优化,共掺杂ZnO超晶胞模型如图1所示,此时形成浓度为12.5%的共掺杂ZnO结构,其中选取了Co和Y的3种配位进行了对比计算,分别为近邻(1),次近邻(2)及远邻(3)。

图1 Co-Y共掺杂ZnO的(2×2×2)超晶胞模型Fig.1 Model of Co-Y co-doped ZnO supercell
本文所有的计算工作都是由基于密度泛函理论(DFT)的CASTEP软件包完成。对电子结构进行计算时采用的是平面波赝势方法,用广义梯度近似(GGA)的PBE泛函处理电子间的交换关联能,选取的价电子组态分别为:Zn3d104s2、O2s22p4、Co3d74s2、Y4s24p64d1。采用4×4×2的Monkhorst-Pack特殊K点对布里渊区积分,能量计算都在倒易空间中进行,平面波截断能为Ecut=400 eV。自洽场计算中,体系能量的收敛精度为1.0×10-5eV/atom,内应力收敛精度为0.05 GPa,作用在每个原子上的受力不大于0.03 eV·nm-1,原子最大位移收敛标准为0.001 nm。在计算之前,先对晶胞结构进行了优化以提高计算精确度。
2 结果与讨论
2.1晶体结构
掺杂前后ZnO超晶胞几何优化后的计算结果如表1所示。本征ZnO超胞在优化后晶格常数a和c分别为0.665和1.059 nm,与袁俊辉等[3]的计算结果相符。在Co单掺入ZnO后晶胞参数和晶胞体积有小幅度的减小,而Y单掺入ZnO会使晶胞参数和晶胞体积较之本征ZnO略有增大,在Co-Y共掺杂ZnO时,晶胞参数和晶胞体积都比本征时略大,处于Co和Y单掺时两者之间,出现这种结果的原因是Y3+、Zn2+和Co3+离子半径的不同(0.09 nm>0.074 nm>0.072 nm)。
表2为优化后ZnO各原子间的重叠集居数,其中已有部分数据取平均值。由表2可知,对于本征ZnO,Zn原子和O原子之间为极性共价键。掺杂使体系的电荷重新分配,可以发现掺杂原子对周围原子的成键有很大影响。从表2可以看出,对整个超晶胞求平均后掺杂体系的Zn-O键的布居数有所减小。由于晶体的各向异性,Co和Y替换Zn后与周围的O发生作用,在不同方向上成键的强弱不同。掺杂体系中,Co-O键和Y-O键与Zn-O键相比,在平行于c轴的方向上,集居数变小,成键的共价性减弱,离子性增强;在垂直于c轴的方向上,集居增加,成键的共价性增强。与单掺杂体系相比,Co-Y共掺杂后,Co原子的集居数变小,Co-O键离子性增加,说明Y的掺入减小了Co周围的电子云密度,对Co离子具有氧化作用。

表1 本征和掺杂ZnO超晶胞优化后的晶格参数Table1 Optim ized lattice constants of intrinsic and doped ZnO supercell

表2 优化后本征和掺杂ZnO各原子间的M ulliken重叠集居数Table2 M ulliken bound populations of intrinsic and doped ZnO after optim ization
2.2能带结构与态密度分析
图2对比考察了掺杂前后ZnO的能带结构,图中只给出了费米能级(0 eV处为费米能级)附近(-4~4 eV)的能带结构,以便更加清晰。从图2可以看出,对于本征ZnO,价带顶和导带底都位于G点处,说明ZnO为直接带隙半导体,禁带宽度Eg=0.73 eV。此计算结果与文献[18]得到的结果完全一致,但比实验值(Eg=3.37 eV)偏低。这主要是广义梯度近似(GGA)存在Eg计算值偏低的普遍性问题所致[3]。对于本文计算的本征ZnO结构,由于Zn3d态的能量被过高估计,使得其与O2p态之间的相互作用增强,以致于价带带宽增大,所以计算得到的ZnO带隙宽度偏低。我们更关心的是掺杂带来的变化的趋势及其机理。从图2(b)可以看出,与未掺杂ZnO相比,掺入Co后,会在导带底引入杂质能级,同时在禁带中也出现了杂质能带,费米能级上移,是n型掺杂。从图2(c)可以看出,与未掺杂ZnO相比,掺入Y后,带隙略有减小,费米能级上移进入导带,导带电子浓度增加属于n型掺杂。Co-Y共掺后,导带与部分杂质能级相连,费米能级处的能带结构更加的丰富。其原因是相对于单掺杂情况,共掺杂时的协同效应会使Co离子和Y离子出现更多的价态,费米能级进入导带,半导体发生简并的同时呈现金属性。
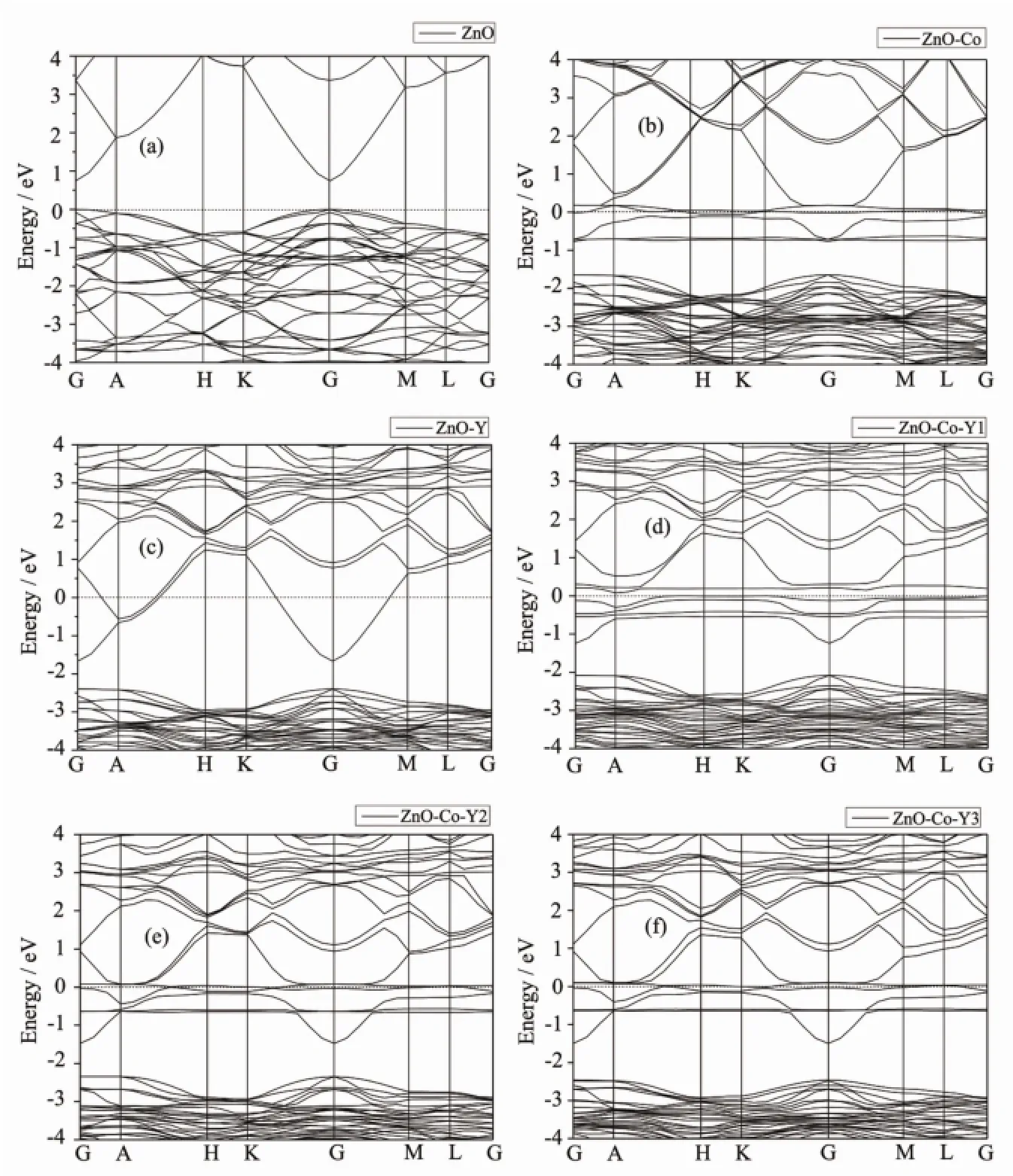
图2 各ZnO体系的能带结构Fig.2 Energy band structures of every ZnO system
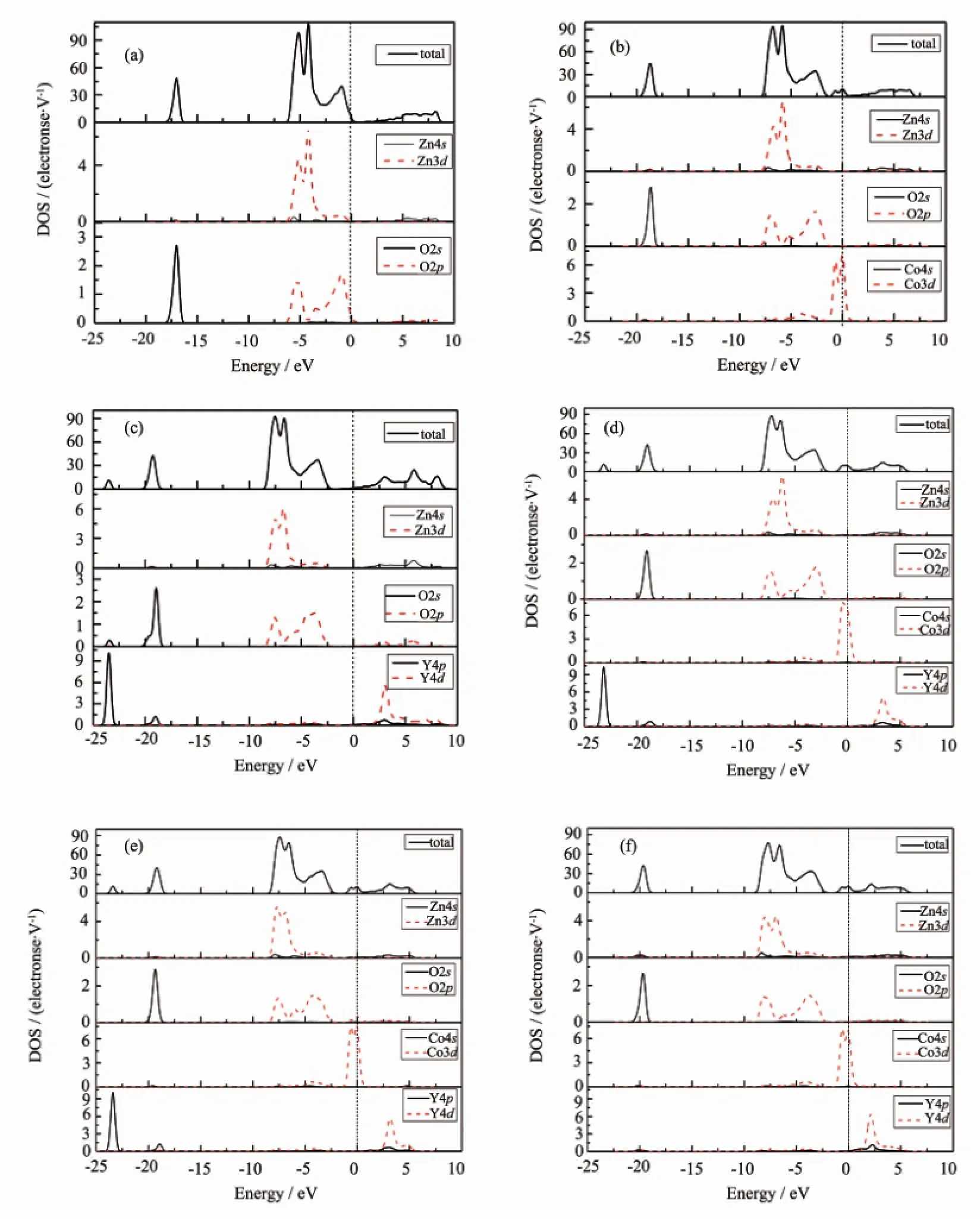
图3各ZnO体系的态密度Fig.3 PDOSof every ZnO system
图3为计算所得本征及Co、Y单原子掺杂和Co-Y共掺ZnO的总态密度(TDOS)图和分波态密度(PDOS)图。从图3(a)中可以看出,对于本征ZnO,其导带部分主要是由Zn4s态提供,而-6~0 eV的价带则是由Zn3d态和O2p态轨道杂化形成,在-18~16 eV处还存在由O2s态形成的孤立能带。从图3 (b)中可以看出,与本征ZnO比较,单掺杂Co时,一部分Co3d态与O2p态和Zn3d态共同构成价带,增加了价带的电子态密度,另一部分Co3d态在禁带中形成杂质能级,有助于电子从价带到导带的跃迁,增加了ZnO的电导率。单掺杂Y时在-23.8~23.0 eV和-19.4~18.6 eV处增加了由Y4p态电子形成的能带,而Y4d态电子主要在导带的低能级区域有所贡献。图3(d,e,f)为3种配位下Co-Y共掺ZnO时的态密度图,可以看到在费米能级附近依然存在着强度有所不同的杂质能带,且Y4d态和Y4p态及Co3d态的作用将会导致体系中电子在各量子态之间的跃迁受到影响。还可以看出,相对于Co单掺杂ZnO,共掺杂体系中Y的掺入使Co离子在费米能级处的电子态密度有一定的增加。在此处由于无论对ZnO晶体的价带还是导带,Y的s轨道电子态密度都只有很小的贡献,并且只作用在更深能级处(<-40 eV),所以在此处可以忽略不计。通过以上分析,过渡金属Co、Y替换Zn掺杂ZnO后会导致能带数目增多,并且对ZnO掺杂结构的性质起着主要作用是其d态电子。
2.3光学性质
在第一性原理研究中,材料的光学性质主要是通过介电函数来研究。在线性响应范围内,固体的宏观光学响应函数通常由光的复介电函数(ω)进行描述:

其中ε1=n2-k2,ε2=2nk分别为介电函数的实部与虚部,ω是频率。介电函数的虚部ε2(ω)与光学吸收直接相关,它可以由电子跃迁的选择定则,占据态的电子波函数以及未占据态的电子波函数之间的矩阵元计算从而得出。根据Kramers-Kronig色散关系和直接跃迁概率的定义可以推导出晶体介电函数的实部ε1、虚部ε2、吸收系数α(ω)、反射系数R(ω)和能量损失谱L(ω)等。在这里直接给出结果[18]:
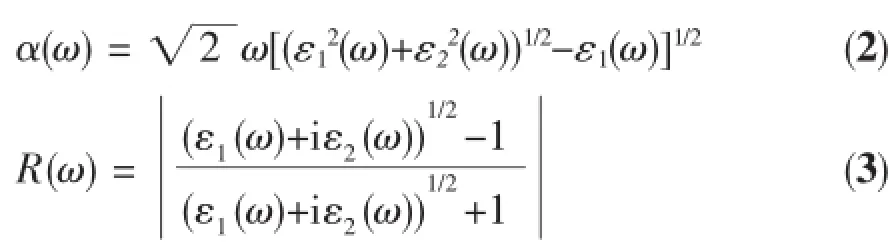
由于密度泛函理论计算所得到的带隙通常会比实验值小,因此本文在计算光学性质时用剪刀算符对带隙进行校正。剪刀算符取ZnO禁带宽度的实验值(Eg=3.37 eV)与ZnO禁带宽度的计算值(Eg= 0.74 eV)之差2.63 eV。垂直入射光的极化方向为<100>方向。
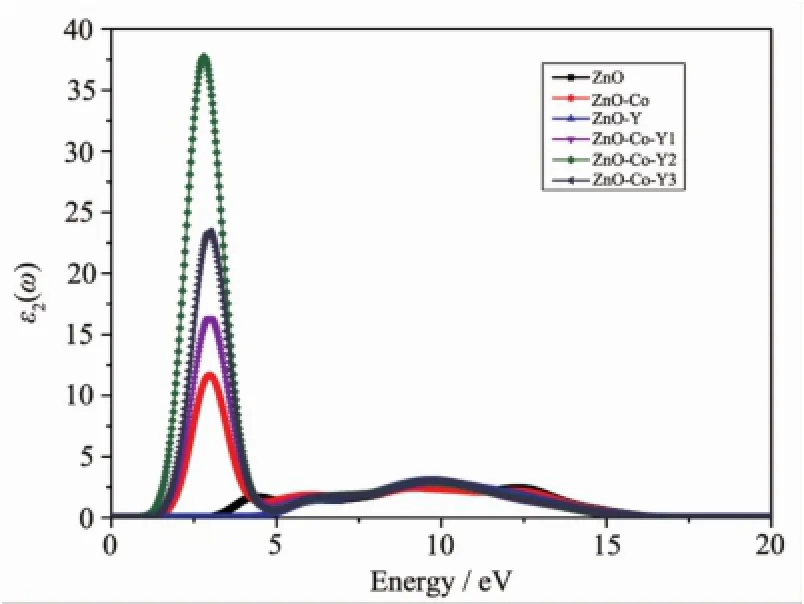
图4本征ZnO和各掺杂ZnO的介电函数虚部Fig.4 Imaginary part of dielectric function of intrinsic and doped ZnO
图4为本征ZnO及各掺杂ZnO介电函数虚部。从图4可以看出,本征ZnO的介电谱存在3个峰,分别位于4.4,9.1和12.3 eV处,与文献[3]计算得到的结果(4.3,9.1,12.6 eV)十分接近。由对ZnO的能带结构和分态密度的分析可知,第一个峰主要来源于价带附近的O2p态电子与导带底附近的Zn4s态电子之间的跃迁,第二个峰主要是由Zn3d态和O2p态电子之间的跃迁引起,第三个峰则是由价带中Zn3d态和O2s态之间的跃迁所致。Co单掺杂后主要存在4个峰,分别位于3.0、6.1、9.2、12.2 eV附近,Y单掺杂后则出现了3个峰,分别是在6.1、9.7、12.5 eV附近。Co掺杂主要引入了Co3d态电子与O2p态电子之间的跃迁,对应3.0 eV附近的峰值,而Y掺杂则主要引入Zn3d态与Y4d之间的跃迁,对应9.7 eV附近的峰值。在共掺杂体系当中,Co的离子性更强,因此3.0 eV附近的峰值更大,这与前面的重叠集局数分析结果相符。可以发现,ZnO光学性质受掺杂影响较大的主要是能量较低的区域,而较高能量区域的变化相对而言要小很多。这主要归因于Co3d态电子与导带底附近O2p态电子之间的跃迁。
图5为本征及各掺杂ZnO的吸收系数随光子能量变化的曲线,从图5中可以看出,在Y掺杂ZnO后,体系的吸收边出现蓝移,且在高能出现吸收峰,使得ZnO在紫外区域的吸收增强,与实验得到的结果相符[20]。而当Co单掺入ZnO后,体系的吸收峰则出现明显的红移,使得ZnO近紫外区域的吸收边向长波方移动,从图5中可以看出,在Co单掺入ZnO后会使得体系在可见光区域的吸收系数增强,而在Co-Y共掺后,低能区域的吸收峰值进一步增加,同时高能区域的吸收峰明显红移并且展宽,这主要是载流子浓度增加导致的[3]。由上述对态密度的分析可知,共掺体系中Co离子在在费米能级处的电子态密度会进一步增加,而Co3d态电子与O2p态电子之间的跃迁恰好增加了可见光区域的光吸收系数。相对于单掺杂体系,Co-Y共掺杂后对可见光波段的吸收能力有较大的增加,有利于在光电感应器件中的应用。
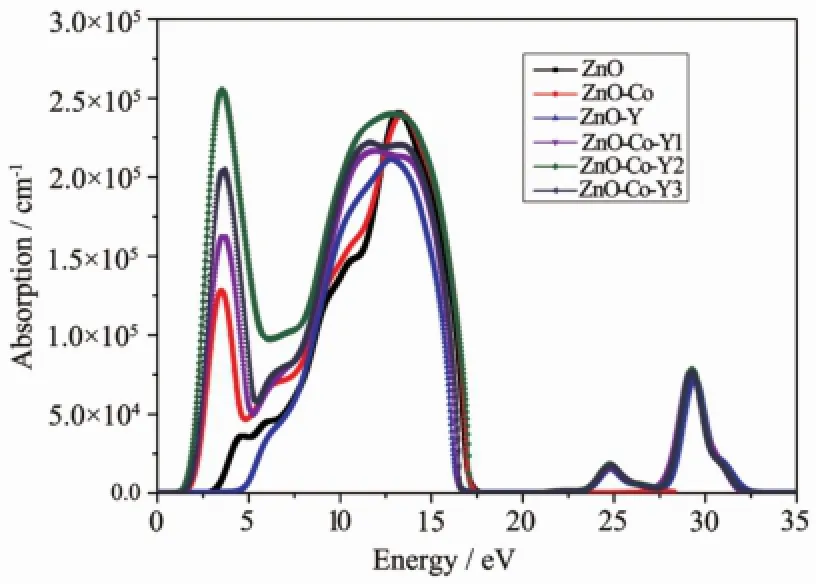
图5 本征ZnO及各掺杂ZnO的吸收谱Fig.5 Absorption spectra of intrinsic and doped ZnO
图6为本征和各掺杂ZnO的反射谱。从图6中可以看出,Y单掺ZnO时,体系的反射率与本征ZnO相比在80~250 nm波段略有增大,其余部分均低于本征ZnO。在Co单掺杂时,体系的反射率在低能方向上明显增强,在高能方向也略有增强。而在Co-Y共掺体系中可以发现对于高能区域和低能区域,反射率都有较大的增强。

图6 本征ZnO及各掺杂ZnO的反射谱Fig.6 Reflectivity spectra of intrinsic and doped ZnO
3 结论
本文通过第一性原理平面波赝势法建立了本征ZnO、Co和Y单掺杂ZnO、Co-Y不同配位共掺杂ZnO的超胞模型,并分别对模型进行了几何结构优化、能带结构分布、态密度分布和光学性质的计算。计算结果表明,掺杂后ZnO中平行于c轴方向的共价键减弱,垂直于c轴方向的共价键增强。与Co单掺杂体系相比,Co-Y共掺杂ZnO的Co原子的集居数变得更少,表明Y的掺入对Co离子具有氧化作用;通过对能带图的分析可以发现,在ZnO中,单掺Co和Y都可以提高ZnO中载流子浓度,从而提高ZnO的导电性。光学计算则表明,在Co-Y共掺杂ZnO体系中,Y离子的引入,一方面增强了体系在紫外区域的吸收,另一方面使Co3d态电子在费米能级附近的2个电子密度峰距离更近,使得共掺体系的光子能量吸收在可见光区域大幅增加,同时在近紫外区域红移并展宽,因此Co-Y共掺杂ZnO在光电感应器件和太阳能电池的工业生产中具有广阔的应用前景。
[1]CHEN Xi(陈熙),LI Li(李莉),ZHANGWen-Zhi(张文治), et al.Chinese J.Inorg.Chem.(无机化学学报),2015,31(10): 1971-1980
[2]WANG Zi-Fang(王志芳),LIMi(李密),ZHANG Hong-Xia (张红霞),et al.Chinese J.Inorg.Chem.(无机化学学报), 2012,28(4):715-720
[3]YUAN Jun-Hui(袁俊辉),GAO Bo(高博),WANG Wen (汪文),etal.Acta Phys.-Chim.Sin.(物理化学学报),2015, 31(7):1302-1308
[4]HE Jing-Fang(何静芳),WU Yi(吴一),SHI Ru-Qian (史茹倩),et al.Chinese J.Inorg.Chem.(无机化学学报), 2015,31(1):15-22
[5]GAO Li(高立),ZHANG Jian-Min(张建民).Acta Phys.Sin. (物理学报),2010,59(2):1263-1267
[6]Mass J,Bhattacharya P,Katiyar RS.Mater.Sci.Eng.,2003, B103:9-15
[7]Limaye M V,Singh SB,Das R,et al.J.Solid State Chem., 2011,184:391-400
[8]Chattopadhyay S,Neogi SK,Sarkar A,et al.J.Magn.Magn. Mater.,2011,323:363-368
[9]XIAO Zhen-Lin(肖振林),SHI Li-Bin(史力斌).Acta Phys. Sin.(物理学报),2011,60(2):027502(8Pages)
[10]GUO Shao-Qiang(郭少强),HOU Qing-Yu(侯清玉),ZHAO Chun-Wang(赵春旺),et al.Acta Phys.Sin.(物理学报), 2014,63(10):107101(7Pages)
[11]LIU Wei-Jie(刘玮洁),SUN Zheng-Hao(孙正昊),HUANG Yu-Xin(黄宇欣),etal.Acta Phys.Sin.(物理学报),2013,62 (12):127101(7Pages)
[12]BAO Xiu-Li(包秀丽),LIN Lin(林琳).J.Atom.Mol.Phys. (原子与分子物理学报),2015,32(3):487-491
[13]WU Yan-Nan(吴艳南),WU Ding-Cai(吴定才),DENG Si-Hao(邓思浩),et al.J.Sichuan Normal Univ.(四川师范大学学报),2012,35(1):95-100
[14]QU Sheng-Wei(曲盛薇),TANG Xing(唐鑫),LV Hai-Feng (吕海峰),et al.Chin.J.Lumin.(发光学报),2010,31(2): 204-208
[15]Heo S,Sharma SK,Lee S,et al.Thin Solid Films,2014,558: 27-30
[16]Zheng J H,Song J L,Jiang Q,et al.Appl.Surf.Sci., 2012,258:6735-6738
[17]WU Ding-Cai(吴定才),HU Zhi-Gang(胡志刚),DUAN Man-Yi(段满益),et al.Acta Phys.Sin.(物理学报),2009,58(10): 7261-7266
[18]HE Jing-Fang(何静芳),ZHENG Shu-Kai(郑树凯),ZHOU Peng-Li(周鹏力),et al.Acta Phys.Sin.(物理学报),2014, 63(4):046301(7Pages)
[19]Anandan S,Muthukumaran S,Ashokkumar M.Superlattice Microstruct.2014,74:247-260
[20]Anandan S,Muthukumaran S.Opt.Mater.,2013,35:2241-2249
First-Princip les Calculations of the Electronic Structure and Optical Properties of Co-Y Co-doped ZnO
FAN Tao-Jian YUAN Jun-Hui YANG Yong-Yong YU Nian-Nian*WANG Jia-Fu*
(School of Science,Wuhan University of Technology,Wuhan 430070,China)
The electronic structures and optical properties of intrinsic Co,Y doped and Co-Y co-doped ZnO compounds are calculated using the first principle pseudopotentialmethod of plane wave based on the density functional theory.The results show that the conductivity of ZnO can be improved by Co and Y doping because of the increase in carrier concentration under the order ofmagnitude of the doping concentration in this paper.Co-Y co-doping leads to degeneration and makes ZnO metallic.Co-doped ZnO can show enhanced light absorption in the visible and near ultraviolet regions,while doping with Y enhances absorption in the ultraviolet region.Co-Y co-doping greatly increases the absorption of visible and near ultraviolet regions because of the synergistic effect between Y ions and Co ions,which can be used to prepare the opto-electronic devices.
ZnO;rst-principles;electronic structure;optical properties
O649.4
A
1001-4861(2016)07-1183-07
10.11862/CJIC.2016.162
2016-02-25。收修改稿日期:2016-05-22。
国家自然科学基金(No.11504281)资助项目。
*通信联系人。E-mail:jasper@whut.edu.cn,niannianyu@whut.edu.cn;Tel:+86-13971473049,+86-15926485056
