原位合成石墨烯负载的Co-P催化剂及光解水制氢
2016-12-05郭跃萍吕功煊
郭跃萍 吕功煊
(1中科院兰州化学物理研究所,羟基合成与选择氧化重点实验室,兰州730000) (2兰州大学化学化工学院,兰州730000)
原位合成石墨烯负载的Co-P催化剂及光解水制氢
郭跃萍1,2吕功煊*,1
(1中科院兰州化学物理研究所,羟基合成与选择氧化重点实验室,兰州730000) (2兰州大学化学化工学院,兰州730000)
在石墨烯(GP)基底上利用光电子原位还原合成了具有非晶态结构的Co-P/GP光催化剂。通过X射线衍射仪(XRD)和透射电子显微镜(TEM)表征,证明所制备的催化剂在石墨烯载体上分散性好、粒径小,具有非晶态结构。由XPS表征了催化剂的表面电子状态,发现催化剂表面的Co和P主要是以还原态存在,有利于表面富集电子。经过曙红敏化,催化剂在可见光辐照下具有很好的光催化制氢性能。考察了不同物质的量之比nCo∶nP的制氢速率,当nCo∶nP=5∶1时,3 h的平均制氢量为24.5mmol·h-1·gCo-1。考察了不同波长条件下催化剂制氢的量子效率,当波长为430 nm时,Co-P/GP催化剂的制氢量子效率AQE达到8.4%,比没有添加P的Co/GP催化剂提高了1.5倍。
非晶态合金;光催化;原位合成;石墨烯;制氢
随着石化能源的消耗和温室气体的大量产生,人们对清洁能源的需求越来越迫切[1-5]。氢气作为一种可再生的清洁能源被科学家广泛研究,而在这些研究中,利用容易得到的太阳能来制备可储存的氢气显得非常有意义。光解水制氢技术作为一种直接利用太阳能,并将其转变为清洁的二次能源,成为近年来的研究热点[6-10]。
由于石墨烯具有特殊的二维结构以及其类金属的良好电子传导性,使其非常有利于光生电子、空穴的分离和迁移,提高其量子效率。所以在光解水制氢的研究中,作为电荷传输和分离的材料,石墨烯很受关注[11-19]。同时石墨烯特殊的层状结构还可以使负载于其上的催化剂颗粒限域生长,颗粒趋于细化[20]。且其大的比表面积和特殊的官能团可使负载其上的催化剂颗粒均匀分布,减少团聚[21]。现阶段光解水制氢的研究中选取的助催化剂一般为贵金属催化剂如Pt[22]、Pd[23]、Ag[24]等,但这些贵金属在地壳中含量很低,不适合大规模制氢,故需要开发代替贵金属的助催化剂。Matthew等[25-26]在nCo∶nP=1∶2的含有磷酸盐的缓冲溶液中,通过电还原的方法原位合成了具有类似光合作用的Co-P催化剂,考察了其光解水产氧的效率。发现磷酸盐阴离子在此人造光合体系中起到了接受水裂解质子的作用,促进了光合循环中电子、质子的分离和传递。另外,Patel等[27]认为将P加入Co基催化剂中可以形成掺杂的Co-P合金催化剂,由于其具有特殊的非晶态结构,使得表面具有较多的配位不饱和键,容易吸附反应物分子,在作为催化剂使用时具有很高的催化效果。基于此,Co基催化剂被广泛应用于加氢催化剂的研究中,并得到了很高的效率[28-31]。故将此类过渡金属非晶态合金引入作为光解水制氢的助催化剂,期望得到可以替代贵金属助催化剂的制氢体系,降低大规模光解水制氢的成本。
本文采用光生电子还原金属离子,并将其负载于石墨烯上,原位合成金属磷化物/石墨烯的光催化剂,并将其用于光解水制氢的反应中。选取荧光素染料曙红为光敏化剂,三乙醇胺为电子牺牲剂,在可见光条件下考察其制氢性能。
1 实验部分
1.1试剂和仪器
实验中所用的化学试剂除石墨粉(Graphite)外都为分析纯,有五氧化二磷(P2O5)、过硫酸钾(K2S2O8)、浓硫酸(H2SO4)、水合肼(N2H4·H2O)、硫酸钴(CoSO4·H2O)、次磷酸钠(NaH2PO2·H2O)、三乙醇胺(TEOA)、曙红(EY)等。
光还原以及光解水制氢所用的电源都为300 W氙灯,并配有420 nm截止滤光片。催化剂结构用型号为Rigaku D/max-2400的X射线粉末衍射仪(XRD)分析,用Cu靶作为射线源,Kα射线,λ= 0.154 187 nm,电压40 kV,电流100 mA,步长为0.02°,扫描角度为10°~70°。催化剂形貌在Tecnai F-30 FEG型高分辨透射电镜上分析。表面电子状态用PHI-5702型多功能X射线光电子能谱仪(XPS)测量。制氢过程中的氢气用Agilent6820 GC8800型气相色谱检测。
1.2载体石墨烯的制备
实验中采用改进后的Hummers法制备氧化石墨(GO),采用水合阱还原得到石墨烯(GP)。具体过程如下:将10 g Graphite、5 g P2O5和5 g K2S2O8加入到50 mL浓硫酸中,剧烈搅拌,将温度升至85℃,反应5 h。冷却至室温,用蒸馏水稀释,并过滤、洗涤至中性,得到GO。将得到的GO稀释到蒸馏水中,标定至2mg·mL-1。为了保证还原效果,需进一步稀释GO溶液。取50mLGO溶液,稀释至200 mL,油浴中升温至95℃,用浓氨水将溶液调至pH=10,在搅拌条件下加入85μL水合肼[32],反应1 h。冷却至室温,标定得到0.4 mg·mL-1GP溶液,用于后续实验中。
1.3催化剂制备及光解水制氢过程
实验中所用的催化剂采用原位还原的方法生成,具体过程如下:在带有硅胶塞的密闭石英反应瓶中加入80 mL三乙醇胺(TEOA,10%,V/V,pH= 10),15 mL的GP溶液(0.4 mg·mL-1),超声15 min。加入2mL配置好的Co和Co-P(nCo∶nP为6∶1,5∶1,1∶1,1∶10)溶液,活性位溶液的配比如表1所示,其中柠檬酸钠作为配位剂使用。加入56mg曙红染料,超声15min,氩气排气30 min。在搅拌条件下使用300W氙灯并加420 nm截止滤光片光照还原、制氢,光照时间为3 h。循环实验是将每次反应完成后的催化剂用离心机分离,再加入新鲜的TEOA和EY,继续光照反应,循环4次。
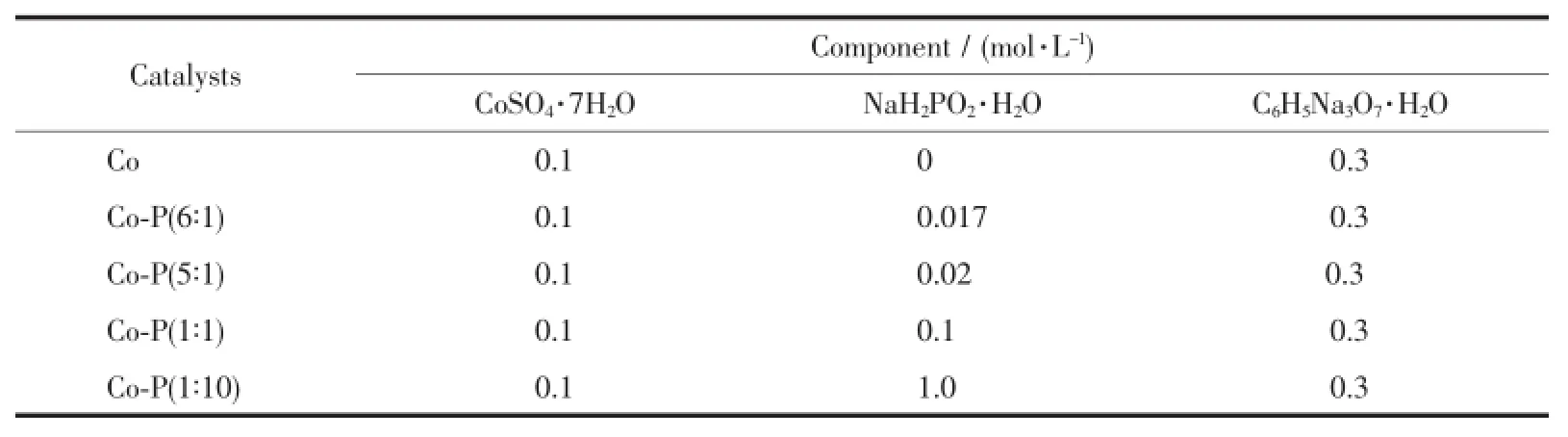
表1 活性位溶液配比表Table1 Bath com position of the catalysts
1.4量子效率的测量以及计算
采用与制氢实验相同的实验条件,用430、460、480、500、520、540 nm的滤光片滤去其它波长的光,光照反应1 h。采用FU 100型辐射计测定入射光子数,采用公式(1)计算各波段下的表观量子效率。

2 结果与讨论
2.1样品表征
图1是光还原得到的Co/GP和Co-P/GP(nCo∶nP= 5∶1)的催化剂XRD图。从图中可以看到,在25°左右出现的峰为石墨烯的特征峰。在45°左右出现的宽化的峰可归为催化剂的衍射峰。从图中可以发现得到的合金催化剂结构接近非晶态,而由于Co-P/GP催化剂的衍射峰更加宽化,所以其非晶化程度更高,说明添加P会使合金催化剂的结构向非晶态转变。

图1 Co/GP、Co-P/GP催化剂的XRD图Fig.1 XRD patterns of Co/GP and Co-P/GP
图2(a)是nCo∶nP=5∶1时Co-P/GP催化剂的TEM图,从图中可以看到催化剂颗粒沉积在石墨烯表面,但是观察不到明显的颗粒。图2(b)是HRTEM图,从图中可以发现石墨烯表面上负载有粒径为3~ 4 nm的催化剂颗粒。用EDX定性分析催化剂表面成份,可以发现其中有C、O、Br、P和Co成份,见图2(c)。图中的Cu峰来源于测量时的Cu网,Br峰来源于残留的EY。而Co和P的峰则说明活性位溶液中的Co2+和H2PO2-被光生电子还原形成Co-P合金。在反应体系中加入的Co2+和H2PO2-被石墨烯表面的羟基吸附,而配位剂离子的作用是将其还原电位拉近。溶液中的EY吸收可见光的光子,产生激发态的EY1*和EY3*,EY3*被电子牺牲剂EDTA还原成EY-,而EY-具有-0.8 V的氧化还原电位[33],所以可以将配位在石墨烯表面的Co2+和H2PO2-还原形成Co-P合金,由于光生电子分布均匀且还原温和,故可形成均匀的非晶态合金颗粒。
图3是nCo∶nP=5∶1时的Co-P/GP催化剂的X射线光电子能谱分析图。从图3(a)中可以看出,得到的催化剂表面的主要成份是Co、P、C和O。C峰是由石墨烯载体产生,O峰是样品在等待测试的过程中被氧化所致。由于Co、P峰信号很弱,故对其精细峰进行了分析。

图2 Co-P/GP(5∶1)催化剂的TEM(a)、HRTEM(b)和EDX(c)图谱Fig.2 TEM(a)and HRTEM(b)images of Co-P/GP catalysts and EDX spectrum(c)of Co-P/GP catalysts(precursor:nCo∶nP=5∶1)

图3(a)Co-P/GP催化剂的X射线光电子能谱分析(a)全谱,(b)Co2p3/2和(c)P2pFig.3 XPS survey spectra of Co-P/GP(a),Co2p3/2(b)and P2p(c)
图3(b)是Co2p3/2轨道下的精细峰,从图中可以发现,在779.1和784.0 eV出现的峰分别是Co2p3/2的束缚态和氧化态(Co2+)的峰[34]。而由于779.1 eV的峰面积(59.3%)比784.0 eV的峰面积(40.6%)大,说明催化剂中Co的基态占多数。这将有利于催化剂表面富集电子,促使H+得电子,加快反应中H2的生成。由于催化剂在制备过程中加入的P很少导致在(c)图中P的信号峰很弱,但是经过分析发现在130.4 eV出现的是P2p3/2束缚态[35]的峰,说明溶液中的有一部分H2PO2-被还原形成基态的P。
2.2光解水制氢性能
图4(a)示出了所得催化剂在EY敏化下,在pH=11时的TEOA(10%,V/V)中,催化还原水制氢的动力学曲线图。从图中可以发现,当催化剂体系中不含P时,光解水制氢速率较慢,在3 h内产生氢气667μmol,若以制氢活性位Co的克数计的话,其3 h平均制氢速率为18.8mmol·h-1·gCo-1。在催化体系中加入P后,催化剂的制氢速率提高,虽然P的加入量对催化制氢的影响不大,但是在nCo∶nP=5∶1时得到了3 h最大制氢量为872μmol,以制氢活性位Co的克数计的话,其3 h平均制氢速率为24.5 mmol·h-1·gCo-1。图4(b)示出了以活性位Co计的3 h平均制氢速率,由图中可以看出本体系最合适的nCo∶nP为5∶1。Min[36]等认为有石墨烯载体的催化剂具有高的光解水制氢效率,主要是由于石墨烯载体可以快速的分离光生电子-空穴对,并大幅降低其复合速度,使得催化活性位表面具有较多电子。溶液中被敏化的EY3*被电子牺牲剂EDTA还原成EY-,EY-将电子通过容易传导的石墨烯表面传递到活性位Co的表面[36-37],H+在Co表面得电子形成H-H键。而P的存在使得Co活性位表面更富有电子[37],这将大大提高水中的H+吸附和得电子,加快产生H2的速度。
2.3催化剂负载量影响

图4(a)所得催化剂制氢动力学图;(b)3 h平均制氢速率图Fig.4 Kinetic curves of photocatalytic H2evolution from water over the catalysts(a)and the rate of the H2evolution in 3 h(b)
图5所示为活性最高时的Co-P(5∶1)活性位溶液的加入量对制氢总量影响对比图,反应时间为3 h。从图中可以发现,负载量对制氢活性的影响较大。随着活性位溶液体积的增加,3 h的总制氢量增加。当加入的溶液体积进一步增加时,制氢量反而降低。当加入体积为2 mL时,制氢量达到最大,此时的负载量为197%。说明该反应并不是活性位控制的催化反应,存在最佳的负载量。当负载量较低时,由于活性位不足,通过石墨烯传递的电子不能充分转移到H+得电子的表面;而当活性位溶液加入太多时,容易造成催化剂团聚,致使部分催化剂不能与石墨烯、染料以及电子牺牲剂构成协同反应体系,所以反应时需要选取合适的负载量。

图5 加入的Co-P(5∶1)溶液量对光催化制氢量的影响对比图Fig.5 H2production comparison of the catalystswith different Co-P solution volumes(precursor: nCo∶nP=5∶1)
2.4量子效率测量
图6是通过测量不同单波长(430、460、480、500、520、540 nm)光照1 h时Co/GP和Co-P/GP(5∶1)催化反应体系的制氢量,并通过公式(1)计算其量子效率的对比图。从图中可以发现,在波长为430 nm时,Co/GP和Co-P/GP催化剂的量子效率最高。其中Co-P/GP催化剂的量子效率达到8.4%,高于CoSx/ GP催化剂在430 nm时的6.7%[19]。

图6 Co/GP和Co-P/GP(5∶1)催化剂制氢的表观量子效率图Fig.6 Apparent quantum efficiencies(AQEs)of H2evolution for Co/GPand Co-P/GP catalysts (precursor:nCo∶nP=5∶1)
2.5催化剂耐久性测试
图7是活性位溶液nCo∶nP=5∶1时得到的Co-P/ GP催化剂的耐久性测试图。从图中可以发现第2次循环时催化剂的制氢量下降很多,其原因主要是由第一次反应完成后离心分离催化剂时细小的Co活性位的损失造成。当循环到第3次和第4次时,催化剂的活性基本保持不变。

图7 Co-P/GP催化剂循环制氢图Fig.7 Stability testing and the compare of H2evolution over the Co-P/GP catalysts
3 结论
在本工作中,通过光生电子还原得到了非晶态的Co/GP和Co-P/GP催化剂。通过可见光区域下的光敏化制氢性能的研究,找到了P掺杂的合适浓度和催化剂的最佳负载量。对所得催化剂在不同光波波长下的量子效率进行了研究,在430 nm时得到了最高的量子效率8.4%。在对催化剂的循环性能的分析中,经过3次循环后催化剂的性能保持稳定。用具有高制氢效率的非晶态P化物代替贵金属催化剂,为光解水制氢技术开发了高效、廉价且能广泛应用的助催化剂,并为其大规模的应用提供了实验基础。
[1]Cortright D,Davda R,Dumesic A.Nature,2002,418:964-966
[2]Zhen W L,Li B,Lu G X.Chem.Commun.,2015,51(9):1728 -1731
[3]HU Xun(胡勋),ZHANG Li-Jun(张丽君),LÜGong-Xuan(吕功煊).J.Mol.Catal.(China)(分子催化),2015,29(5):431-440
[4]Awad H,Veziroglu N.Int J.Hydrogen Energy.,1984,9(5):355-366
[5]PENG Shao-Qin(彭绍琴),DING Min(丁敏),YITing(易婷), et al.J.Mol.Catal.(China)(分子催化),2014,28(5):466-473
[6]LICao-Long(李曹龙),LEI Zi-Qiang(雷自强),WANG Qi-Zhao(王其召),et al.J.Mol.Catal.(China)(分子催化),2015, 29(4):282-289
[7]YANG Yu(杨俞),XIA Long-Fei(夏龙飞),FAN ZE-Yun(范泽云),et al.J.Mol.Catal.(China)(分子催化),2014,28(2):18-187
[8]LIChao-Long(李曹龙),ZHAO Yu-Ting(赵宇婷),CHAO Fei (曹菲),et al.Chinese J.Inorg.Chem.(无机化学学报),2013, 12:2535-2542
[9]MU Jing(穆劲),CHEN Li-Li(陈丽莉),KANG Shi-Zhao(康诗钊),et al.Chinese J.Inorg.Chem.(无机化学学报),2015, 28:251-256
[10]PAN Neng-Qian(潘能乾),LIAO Jian-Jun(廖建军),LIDan-Hong(李丹红),et al.J.Chinese Ceram.Soc.(硅酸盐学报), 2013,41:1366-1374
[11]Lightcap V,Kosel H,Kamat V.Nano Lett.,2010,10:577-583
[12]Kong C,Li Z,Lu G X.Int.J.Hydrogen Energy,2015,40 (31):9634-9641
[13]Li Z,Kong C,Lu G X.Int.J.Hydrogen Energy,2015,40 (30):9061-9068
[14]Kong C,Li Z,Lu G X.Int.J.Hydrogen Energy,2015,40 (17):5824-5830
[15]Zhang W Y,Kong C,Lu G X.Chem.Commun.,2015,51 (50):10158-10161
[16]Kong C,Min S X,Lu G X.Chem.Commun.,2014,50(66): 9281-9283
[17]Kong C,Min SX,Lu GX.ACSCatal.,2014,4(8):2763-2769
[18]Kong C,Min S X,Lu G X.Chem.Commun.,2014,50(39): 5037-5039
[19]Li Z,Wang Q S,Kong C,et al.J.Phys.Chem.C,2015,119 (24):13561-13568
[20]Zheng X,Xu J,Yan K,et al.Chem.Mater.,2014,26:2344-2353
[21]Kamat V.J.Phys.Chem.Lett.,2010,1:520-527
[22]Zhu M S,Li Z,Xiao B,et al.ACSAppl.Mater.Interfaces, 2013,5:1732-1740
[23]Ren S,Ramchandra T,Andrew L,et al.ACSNano,2014,8: 3490-3497
[24]Yasser A,David B,Carmen V,et al.J.Am.Chem.Soc., 2014,136:1182-1185
[25]Matthew W,Daniel G.Science,2008,321:1072-1075
[26]Matthew W,Yogesh S,Daniel G.Chem.Soc.Rev.,2009,38: 109-114
[27]N.Patel,R.Fernandes,G.Guella,A.Miotello,Appl.Catal. B:Environ.,2010,95:137-143
[28]XUE Jing-Jing(薛晶晶),CUIFang(崔芳),HUANG Zhi-Wei (黄志威),et al.J.Mol.Catal.(China)(分子催化),2013,1:10-15
[29]CHEN Lun-Gang(陈伦刚),LIU Yong(刘勇),LONG Jin-Xin (龙金星),etal.J.Mol.Catal.(China)(分子催化),2014,28(1): 29-38
[30]Guo H Z,Liu X,Hou Y H.J.Power Sources,2014,260:100 -108
[31]Oh T,Kwon S.Int.J.Hydrogen Energy,2013,38:6425-6435
[32]MIN Shi-Xiong(敏世雄).Thesis for the Doctorate of Lanzhou Institute of Chem ical Physics(中国科学院兰州化学物理研究所博士论文).2013.
[33]Kong C,Min S X,Lu G X.Int.J.Hydrogen Energy,2014, 39:4836-4844
[34]Yang G J,Gao DQ,Shi ZH,etal.J.Phys.Chem.C,2010, 114:21989-21993
[35]Ouchene M,Senemaud C,Belin E,etal.J.Non-Cryst.Solids, 1983,625:59-60
[36]Min SX,Lu G X.J.Phys.Chem.C,2012,116:25415-25424
[37]Li H X,Wu Y D,Zhang J,et al.Appl.Catal.A:Gen., 2004,275:199-206
In Situ Synthesis of Co-P/GP Photocatalysts for H2Evolution from W ater
GUO Yue-Ping1,2LÜGong-Xue*,1
(1State Key Laboratory for Oxo Synthesis and Selective Oxidation,Lanzhou Institute of Chemical Physics,Chinese Academy of Science,Lanzhou 730000,China) (2College of Chemistry and Chemical Engineering,Lanzhou University,Lanzhou 730000,China)
Utilizing the reducibility of photogenerated electrons,graphene(GP)supported Co-P/GP catalysts were synthesized via the in situ synthesis strategy.XRD characterizations showed that the synthesized catalystswere amorphous and the TEM showed the catalysts were deposited on the graphene surface homogeneously.The X-ray photoelectron spectroscopy(XPS)of Co-P/GP catalyst indicated that Co and Pwere over Co and P elements the catalysts with.The Co-P/GP catalyst exhibited high photocatalytic activities for hydrogen evolution sensitized by Eosin Y(EY).Photoactivities under differentwavelength were investigated.The highest rate of the H2evolution reached 24.5 mmol·h-1·gCo-1under visible light with wavelength longer than 420 nm.The apparent quantum efficiency(AQE)over Co-P/GP(5∶1)catalystunder 430 nm illuminationswas achieved up to8.4%,which was50% higher than the Co/GP catalyst.
amorphous alloy;photocatalyst;in situ synthesis;graphene;hydrogen evolution
O611.3
A
1001-4861(2016)07-1177-06
10.11862/CJIC.2016.152
2015-12-23。收修改稿日期:2016-05-17。
国家自然科学基金(No.21173242)和国家自然科学基金重点项目(No.21433007)资助。
*通信联系人。E-mail:gxlu@lzb.ac.cn,Tel./fax:+869314968178
