针对小鼠Chmp1b基因的可诱导shRNA表达转基因载体的构建及功能鉴定
2016-12-01蔡莹刘柳连博文陈澄
蔡莹,刘柳,连博文,陈澄
(中国医科大学基础医学院发育细胞生物学教研室,教育部医学细胞生物学重点实验室,卫生部细胞生物学重点实验室,沈阳 110122)
针对小鼠Chmp1b基因的可诱导shRNA表达转基因载体的构建及功能鉴定
蔡莹,刘柳,连博文,陈澄
(中国医科大学基础医学院发育细胞生物学教研室,教育部医学细胞生物学重点实验室,卫生部细胞生物学重点实验室,沈阳 110122)
目的设计构建针对小鼠Chmp1b基因的可诱导shRNA表达转基因载体,以用于制备相关转基因小鼠,并在体外验证其有效性。方法剔除pcDNA3.1载体的CMV启动子以及多克隆位点,然后接入带有LacZ⁃fLoxP插入失活的小鼠U6启动子并引入适当的酶切位点,装入设计好的shRNA片段,测试shRNA抑制Chmp1b表达的效率与专一性,检查Cre重组酶介导位点特异性重组去除LoxP位点之间的lac基因后U6启动子活性恢复情况。结果设计的shRNA片段可明显降低Chmp1b的表达,抑制效率达90%以上;成功构建针对小鼠Chmp1b基因的可诱导shRNA表达转基因载体,插入U6启动子中的LacZ⁃fLoxP片段既可以控制U6启动子活性又可以起到报告基因的作用。结论针对小鼠Chmp1b基因的可诱导shRNA表达转基因载体可以用于制备相关转基因小鼠。
shRNA;Chmp1b;Cre/LoxP;转基因载体,构建
网络出版地址
RNA干扰(RNA interference,RNAi)技术提供了一种高效的转录后基因调控方法,可以特异性降解靶基因mRNA[1]。利用这一原理设计的小发卡状RNA分子(shRNA)可以在培养细胞以及模式生物中抑制特定的靶基因表达,抑制效率可以达到90%以上,接近基因敲除水平,也被称为基因敲减[2⁃3]。RNA聚合酶Ⅲ启动子(U6或H1启动子)常被用于细胞或模式生物体内启动shRNA的表达[4]。
Chmp1b(charged multivesicular body protein 1b)又称染色质修饰蛋白,是ESCRT⁃Ⅲ(endosormal sorting complex required for transportⅢ)的相关组成成分之一[5]。真核生物中Chmp1b和ESCRT家族其他成员一起参与多泡体(MVBs)的形成、泛素化标记的膜蛋白分选、胞质分裂等重要细胞生命活动[6]。从小鼠基因组中敲除ESCRT家族成员多导致小鼠胚胎期死亡,影响后续功能基因组学研究[7]。因此诱导条件性基因敲除或敲减是研究ESCRT家族基因功能的必要手段。
相对其他转录后调控手段,shRNA技术因专一性好、操作便捷成为功能基因组学研究的有力工具。为了实现在转基因小鼠体内诱导条件性表达shRNA,本研究参照文献[8]改造了常用的pcD⁃NA3.1质粒,选择lacZ作为报告基因,将带有LoxP侧翼序列的LacZ片段(floxed⁃LacZ)插入小鼠U6启动子,使U6启动子处于失活状态,不能启动shRNA表达。利用Cre重组酶介导的位点特异性重组,将插入小鼠U6启动子中位于2个LoxP位点间的LacZ表达框切除,恢复U6启动子活性,实现针对靶基因shRNA的组织特异性诱导表达,降低靶基因转录,为功能基因组学研究提供有力工具。
1 材料与方法
1.1主要试剂和仪器
pcDNA3.1真核表达载体、Lipofectamine 2000及相关试剂均购于Invitrogen公司;MLE12细胞、限制性内切酶及其他工具酶等购自大连宝生物公司;β⁃tubulin抗体(Abmart公司)、EGFP抗体(天根公司);HRP耦联的抗兔、鼠二抗(中杉金桥公司),1640培养基(Gibco公司)、FBS(Hyclone公司),凝胶成像系统及相关软件为上海天能公司提供;其余为国产分析纯试剂。
1.2shRNA表达载体以及其他研究用载体的构建
本研究所用shRNA表达载体改造步骤简述如下:利用分子克隆技术,自pcDNA3.1载体中去除BglⅡ和BamhⅠ酶切位点间原有的CMV启动子和多克隆位点,去掉pcDNA3.1载体中ApaⅠ酶切位点,设计引物扩增小鼠U6启动子序列通过EcoRⅠ酶切位点装入新载体中,在U6启动子下游引入新的ApaⅠ、XhoⅠ和HindⅢ酶切位点用于后续装入合成的shRNA,该载体命名为pcsh1。在pcsh1载体的U6启动子中剔除-82 bp至-129 bp序列,引入BglⅡ和EcoRⅤ酶切位点,装入含有Sv40 Ori和TK⁃PolyA的LacZ⁃fLoxP序列,该载体命名为pcsh41,是可诱导shR⁃NA表达转基因载体,将用于制备相关转基因小鼠。
参照文献[9]设计了针对小鼠Chmp1bcDNA序列(NM_024190.2)的shRNA序列,核心序列为:5′⁃GGACAAATTCGAACACCAG⁃3′;用作实验对照的scrambleshRNA核心序列为:5′⁃GTAGATGGTCGAC CTTCAC⁃3′。委托上海生工合成由核心序列-茎环序列-核心序列反向互补序列相应酶切位点组成的寡核苷酸片段及其互补序列,退火,并通过ApaⅠ/HindⅢ位点装入pcsh1中构建pcshsic(表达scramble shR⁃NA),pcshcmb7(表达针对小鼠Chmp1b的shRNA)和将针对Chmp1b的shRNA装入pcsh41载体,构建成pcsh41cmb7质粒。pcsh41cmb7质粒在Cre存在的条件下切除floxed⁃LacZ片段,恢复U6启动子活性,表达针对Chmp1b的shRNA。
参照Cre序列设计引物将相应的全长cDNA克隆到pcDNA3.1mychis载体上,表达相应的带有my⁃chis标签的蛋白,质粒命名为pcCre。将psingle质粒(Clonetech,Cat#630933)的EcoRⅠ和XbaⅠ间序列剔除,设计引物扩增EGFP和Chmp1b全长cDNA,装入其中使之表达EGFP⁃Chmp1b融合蛋白,命名为psgCMB01。使用同样的策略构建表达EGFP⁃Chmp1a融合蛋白的质粒psgCMA01。
1.3质粒的共转染及shRNA功能分析
将MLE12细胞以85%汇集度接种到24孔板中,参照Lipofectamine 2000转染试剂手册,将200 ng的psgCMB01质粒分别与600 ng的pcshsic和600 ng的pcshcmb7混合,加入Lipofectamine 2000转染试剂,转染。48 h后检查EGFP荧光,确认针对小鼠Chmp1b的shRNA有效性。利用同样的策略,将pc⁃shcmb7质粒与pcCMB和pcCMA,按1∶2的比例混合共转染MLE12细胞,以pcshsic质粒为对照,Western blotting检查设计的shRNA抑制Chmp1b表达的效率和专一性。
1.4体外Cre介导的位点特异性重组测试
将pcCre质粒用SalⅠ酶切,回收带有Cre表达框的片段,取0.5 μg,按操作手册要求加入20 μL的TNT®Quick Master Mix(Promega,Cat#L1170),1 μL 0.1mol/L的Met,加ddH2O至总体积25 μL,37℃孵育70 min,加入0.1 μg的pcsh41cmb7质粒,继续孵育20 min,取适量反应混合液转化E.coli.DH5α,涂Ampr平板,选单克隆过夜培养,提取质粒,EcoRⅠ酶切鉴定,对选中克隆测序。将得到的正确重组质粒命名为pcshkdcmb7,参照上一步共转染的方法测试pcshkdcmb7抑制Chmp1b表达的能力和专一性。
1.5LacZ细胞化学染色
将MLE12细胞以85%汇集度接种到24孔板中,参照Lipofectamine 2000转染试剂手册,用pc⁃sh41cmb7质粒和pcshkdcmb7质粒转染MLE12细胞,经戊二醛-多聚甲醛固定,Triton X⁃100通透,用X⁃gal染液[0.1 mol/L PBS缓冲液(pH7.3)、5 mmol/L铁氰化钾(K3Fe(CN)6)、5 mmol/L亚铁氰化钾(K4Fe(CN)6·3H2O)、1 mg/mL X⁃gal]染色,观察。
2 结果
2.1诱导表达shRNA的pcsh41cmb7质粒的结构及鉴定
经过复杂的克隆步骤,本研究得到了可诱导表达针对Chmp1b的shRNA的pcsh41cmb7质粒,经酶切鉴定无误。带有LoxP侧翼序列长约3.6×103的 LacZ表达框(floxed⁃LacZ)插入小鼠U6启动子的近端核心序列和远端核心序列之间,使U6启动子处于失活状态,无法表达shRNA分子。如果存在位点特异性重组酶Cre,位于U6启动子中两个通向LoxP位点之间的LacZ表达框被切除,同时保留1个LoxP位点,U6启动子恢复正常的长度(315 bp),该启动子命名为U6⁃kd。见图1。
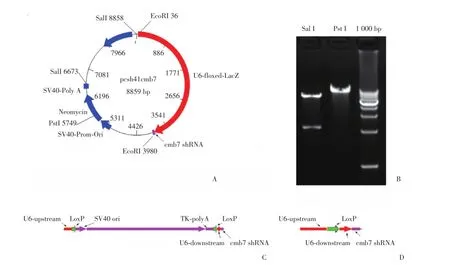
图1 pcsh41cmb7质粒结构及鉴定图Fig.1 Structure and identification map of pcsh41cmb7 plasmid
2.2针对Chmp1b的shRNA可以高效抑制Chmp1b的表达
本研究用pcshcmb7和pcshsic分别与表达EGFP⁃Chmp1b融合蛋白的psgCMB01质粒共转染MLE12细胞。从EGFP⁃Chmp1b融合蛋白免疫印记实验结果可以看到1泳道的pcshcmb7和psgCMB01共转染后EGFP⁃Chmp1b融合蛋白表达量比2泳道的pcsh⁃sic和psgCMB01共转染组降低(图2 A、2B),定量分析显示降低约90%(图2C)。将表达靶基因和报告基因融合蛋白的载体与表达针对该靶基因shRNA的载体共转染细胞测定shRNA的抑制效率是目前比较常用的做法。针对Chmp1b的shRNA可以高效识别EGFP⁃Chmp1b融合蛋白的mRNA中的Chmp1b靶序列,使之被切割降解。
2.3插入的LacZ可使U6启动子失活并具备报告基因功能
MLE12细胞转染pcsh41cmb7质粒并经LacZ染色显示有大量蓝色细胞出现(图3A),说明插入到U6启动子中的LacZ基因具有活性,具有报告基因的功能。MLE12细胞共转染pcsh41cmb7和psgC⁃MB01质粒后检查发现有大量带有GFP荧光的细胞(图3B),说明插入LacZ报告基因后U6启动子失去活性,EGFP⁃Chmp1b融合蛋白表达未被抑制。
2.4通过Cre引发的位点特异性重组恢复了U6启动子的活性
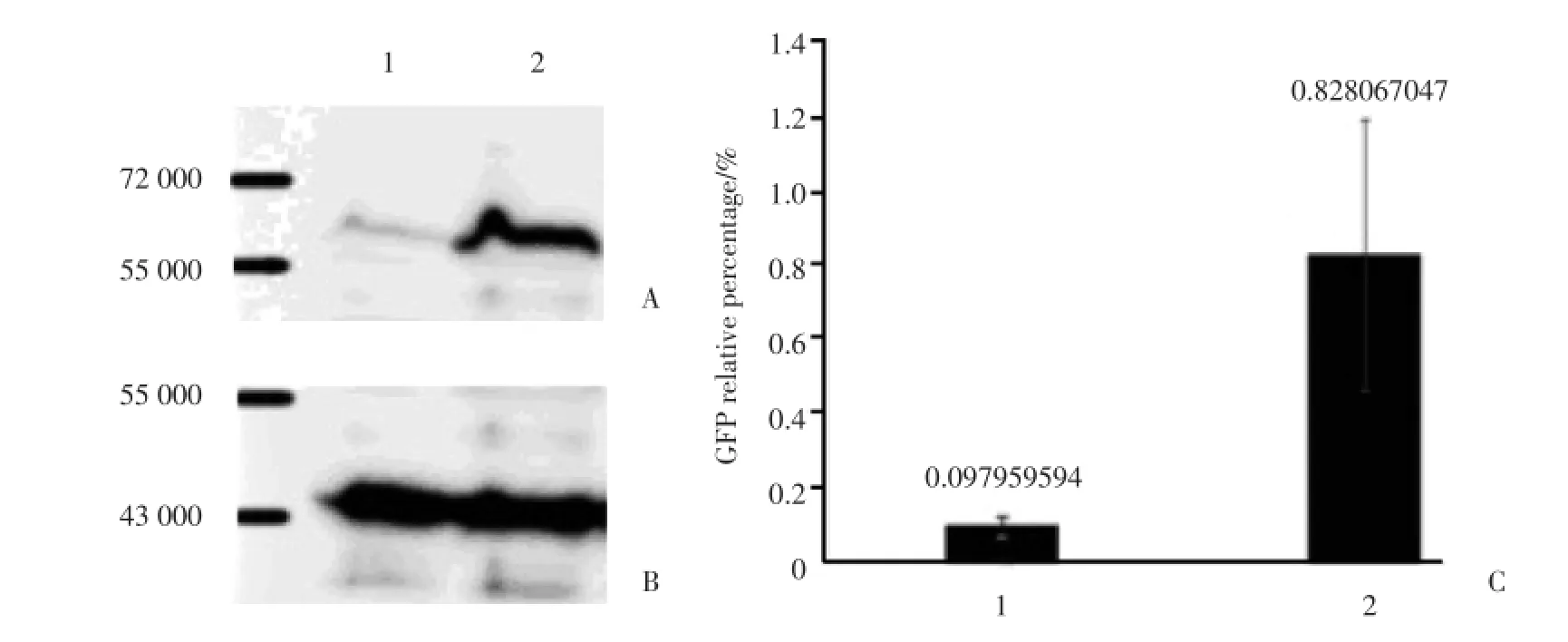
图2 免疫印迹检测针对Chmp1b的shRNA抑制效率Fig.2 The inhibitory effect of shRNA on Chmp1b was detected by Western blotting

图3 报告基因功能显示 ×10Fig.3 Display of the report gene function×10
TNT系统产生的Cre能够介导pcsh41cmb7质粒发生位点特异性重组,用EcoRⅠ酶切鉴定所得重组质粒发现:1,3泳道为正确重组质粒(图4),酶切片段长386 bp,该片段结构如图1D中所示,为U6⁃kd启动子以及其相邻的shRNA,长386 bp,其他为错误重组体。经测序分析确认无误,将其命名为pcshkd⁃cmb7。
本研究发现pcshkdcmb7质粒与psgCMB01共转染的MLE12细胞,与pcshsic与psgCMB01共转染的MLE12细胞相比,带有EGFP荧光表达EGFP⁃Chmp1b融合蛋白的细胞明显减少(图5A、5B),这说明U6⁃kd启动子恢复了U6启动子的活性,由其启动表达的shRNA能引发针对EGFP⁃Chmp1b融合蛋白mRNA的高效切割降解。进一步实验发现pcshkdc⁃mb7质粒与psgCMA01共转染的MLE12细胞也可以看到大量带有EGFP荧光,表达EGFP⁃chmp1a融合蛋白的细胞(图5C),这说明pcshkdcmb7中针对Chmp1b的shRNA序列不能识别同一基因家族的Chmp1a的mRNA序列,具有抑制Chmp1b表达的专一性。
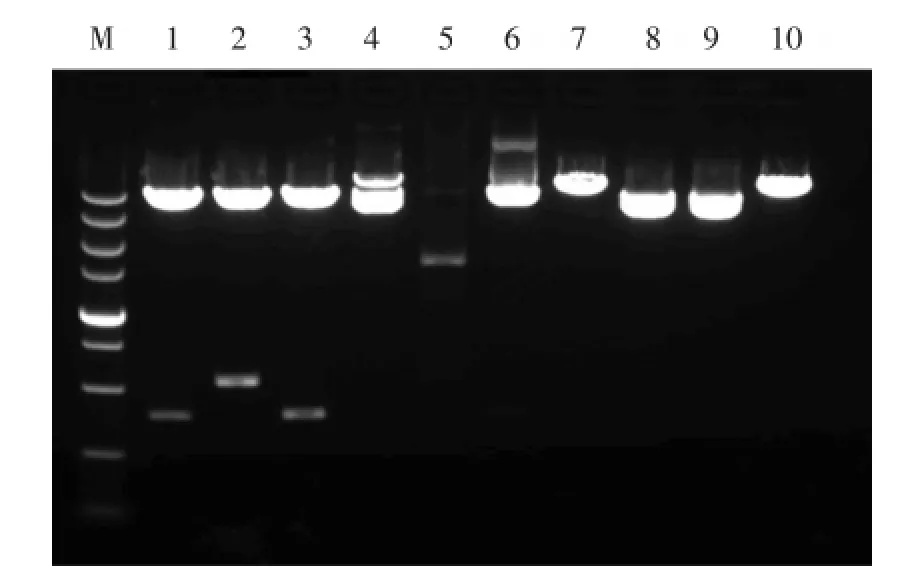
图4 正确重组载体酶切电泳图Fig.4 Correct recombinant vector enzyme electrophoresis
3 讨论
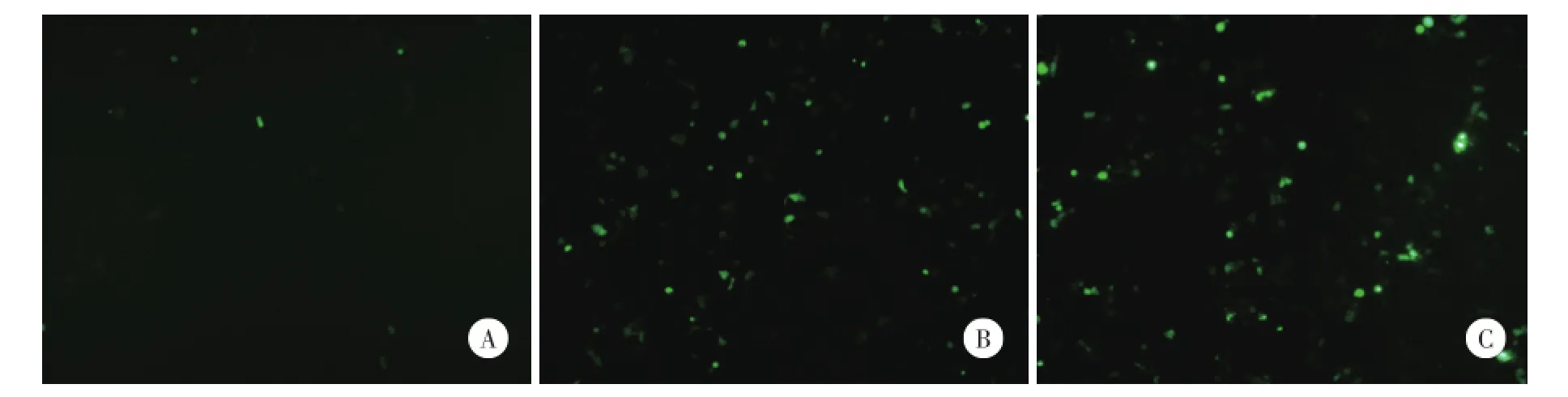
图5 pcshkdcmb7抑制Chmp1b表达的能力 ×10Fig.5 The ability of pcshkdcmb7 to inhibit the expression of Chmp1b×10
目前可以采用3种策略实现可诱导条件性表达shRNA:(1)使用Cre/LoxP系统,通过控制在U6启动子中插入DNA片段与否调控启动子活性[10];(2)使用Cre/LoxP系统通过控制在shRNA茎环结构区插入DNA片段与否控制shRNA结构完整性调控其活性[11];(3)通过改造U6/H1启动子,引入四环素调控元件,达到在小鼠体内实现诱导表达shRNA抑制特定的靶基因表达目的[12⁃13]。其中第2种策略在Cre介导位点特异性重组后残留的LoxP位点可能会影响shRNA的抑制效率和专一性;第3种策略虽然可以实现可诱导shRNA表达,但难以实现组织特异性shRNA表达;而现有的第1种策略可以实现可诱导组织特异性shRNA表达,但是因为没有使用报告基因,不方便追踪小鼠体内发生位点特异性重组表达shRNA的细胞。本研究借鉴了第1种策略思路,将floxed⁃LacZ插入U6启动子中,即能控制U6启动子活性,又可以作为报告基因评估转基因动物靶组织中的shRNA表达的范围。凡是LacZ染色阳性的细胞都是不能表达shRNA的未发生基因敲减的细胞。这为评估可诱导条件敲减转基因鼠靶组织基因敲减范围,追踪相应细胞的增殖分化情况提供了便捷的手段。
为了在体外验证pcsh41cmb7在Cre介导下可否正确重组并恢复U6启动子的活性,本研究利用体外翻译表达系统合成了Cre,加入pcsh41cmb7后得到了pcshkdcmb7质粒,经测序、以及共转染功能检测确认其正确重组且恢复了U6启动子活性,能够表达shRNA抑制包含有Chmp1b序列的融合蛋白的表达。在体外测试时也得到了许多错误重组的质粒,这是由于体外实验存在大量的pcsh41cmb7质粒,Cre介导的位点特异性重组不仅发生在同一个质粒的2个LoxP位点之间,也有可能发生在不同质粒的LoxP位点之间,因此产生了错误重组质粒。但是在体内实验中用该质粒制被转基因动物后,通过有计划地繁殖育种,在转基因动物基因组中可以控制只存在1组pcsh41cmb7插入,此时Cre介导的位点特异性重组被限制在1个经过改造的U6启动子的2个LoxP位点之间,只产生恢复U6启动子活性的正确重组体,不会出现体外实验产生的错误重组体。如果用pcsh41cmb7质粒制备的转基因动物,其体细胞能显示出报告基因LacZ活性。这些体细胞不表达shRNA,未发生基因敲减。而一旦引入Cre,将引发位点特异性重组,切除LacZ,表达针对Chmp1b的shRNA。
综上所述,本研究体外测试的结果证明了可诱导条件敲减质粒pcsh41cmb7可以产生正确且有活性的pcshkdcmb7质粒。将pcsh41cmb7用于后续体内试验是可行的。构建的可诱导条件敲减系统将为快速构建转基因动物,进行功能基因组学研究提供新的更有效的技术手段。
[1]MACRAE IJ,ZHOU K,LI F,et al.Structural basis for double⁃stranded RNA processing by Dicer[J].Science,2006,311(5758):195-198.DOI:10.1126/science.1121638.
[2]PADDISON PJ,CAUDY AA,BERNSTEIN E,et al.Short hairpin RNAs(shRNAs)induce sequence⁃specific silencing in mammalian cells[J].Genes Dev,2002,16(8):948-958.DOI:10.1101/gad.981 002.
[3]VOORHOEVE PM,AGAMI R.Knockdown stands up[J].Trends Biotechnol,2003,21(1):2-4.DOI:10.1016/S0167⁃7799(02)00002⁃1.
[4]MÄKINEN PI,KOPONEN JK,KÄRKKÄINEN AM,et al.RNA in⁃terference:comparison of U6 and H1 promoters in endothelial cellsand in mouse brain[J].J Gene Med,2006,8(4):433-441.DOI:10.1002/jgm.860.
[5]YANG D,RISMANCHI N,RENVOISÉ B,et al.Structural basis for midbody targeting of spastin by the ESCRT⁃III protein CHMP1B[J]. Nat Struct Mol Biol,2008,15(12):1278-1286.DOI:10.1038/ nsmb.1512.
[6]HURLEY JH.ESCRTs are everywhere[J].EMBO J,2015,34(19):2398-2407.DOI:10.15252/embj.201592484.
[7]MICHELET X,DJEDDI A,LEGOUIS R.Developmental and cellu⁃lar functions of the ESCRT machinery in pluricellular organisms[J]. Biol Cell,2010,102(3):191-202.DOI:10.1042/BC20090145.
[8]COUMOUL X,LI W,WANG RH,et al.Inducible suppression of Fg⁃fr2 and survivin in ES cells using a combination of the RNA interfer⁃ence(RNAi)and the Cre⁃LoxP system[J].Nucleic Acids Res,2004,32(10):e85.DOI:10.1093/nar/gnh083.
[9]LI L,LIN X,KHVOROVA A,et al.Definingthe optimal parameters for hairpin⁃based knockdown constructs[J].RNA,2007,13(10):1765-1774.DOI:10.1261/rna.599107.
[10]KANN M,BAE E,LENZ MO,et al.WT1 targets Gas1 to maintain nephron progenitor cells by modulating FGF signals[J].Develop⁃ment,2015,142(7):1254-1266.DOI:10.1242/dev.119735.
[11]KLEINHAMMER A,WURST W,KÜHN R.Constitutive and condi⁃tional RNAi transgenesis in mice[J].Methods,2011,53(4):430-436.DOI:10.1016/j.ymeth.
[12]WANG S,WANG T,JIA L.Type⁃specific and inducible PTEN gene silencing by a tetracycline transcriptional activator⁃regulated short hairpin RNA[J].Mol Cells,2015,38(11):959-965.DOI:10.14348/molcells.2015.0137.
[13]SOCHALSKA M,OTTINA E,TUZLAK S,et al.Conditional knock⁃down of BCL2A1 reveals rate⁃limiting roles in BCR⁃dependent B⁃cell survival[J].Cell Death Differ,2015,23(4):628-639.DOI:10.1038/cdd.2015.130.
(编辑武玉欣)
Construction and Functional Identification of Transgenic Vector Expressing shRNA for MiceChmp1b
CAI Ying,LIU Liu,LIAN Bowen,CHEN Cheng
(Department of Development Cell Biology,College of Basic Medical Science,China Medical University,The Ministry of Education Key Laboratory of Medical Cell Bi⁃ology,The Ministry of Public Health of China Key Laboratory of Cell Biology,Shenyang 110122,China)
ObjectiveTo design and construct a shRNA gene expression vector targetingChmp1bgene mouse,which can be used to prepare transgenic mice,and to verify its effectiveness in vitro.MethodsThe pcDNA3.1 vector CMV promoter and multiple cloning sites were first elimi⁃nated,thenLacZ⁃fLoxPinsertion inactivation of mouse U6 promoter and the appropriate restriction sites and shRNA fragments were introduced into the design.The efficiency and specificity of shRNA for inhibiting Chmp1b expression was tested,the Cre recombinase mediated by site⁃specific re⁃combination LoxP site between lac gene removal was determined,as well as the U6 promoter activity recovery.ResultsExpression of shRNA fragments could significantly reduce Chmp1b expression with an inhibition efficiency above 90%.An inducible shRNA expression transgenic vec⁃tor for mouseChmp1bwas successfully constructed,and insertion of U6 start of fragments LacZ⁃fLoxP can control U6 promoter activity.Conclu⁃sion The constructed shRNA expression vector can be used to prepare transgenic mice.
shRNA;Chmp1b;Cre/LoxP;transgenic vector;construction
Q28
B
0258-4646(2016)11-0968-05
10.12007/j.issn.0258⁃4646.2016.11.003
国家自然科学基金(31271231,30600211)
蔡莹(1987-),女,助理实验师,博士研究生.
陈澄,E-mail:chencheng@mail.cmu.edu.cn
2016-03-03
网络出版时间:
