纳米铝团簇初始氧化过程分子动力学模拟
2016-11-23杨镇何远航
杨镇, 何远航
(北京理工大学 爆炸科学与技术国家重点实验室,北京 100081)
纳米铝团簇初始氧化过程分子动力学模拟
杨镇, 何远航
(北京理工大学 爆炸科学与技术国家重点实验室,北京 100081)
为了研究铝团簇在不同氧化剂中氧化特性及机理,采用ReaxFF反应力场分别对Al-O2和Al-H2O系统的初始氧化过程进行了模拟. 在Al-O2系统中,环境温度决定初始氧化过程铝团簇周围是否出现单独Al原子,且存在一个温度阈值,当低于温度阈值时不会出现单独Al原子. 在Al-H2O系统中,研究了初始水分子数和温度对初始氧化过程的影响. 研究结果表明,水分子对铝团簇表面吸附的水分子离解过程具有促进作用. 分析了直径为2 nm铝团簇与水发生反应的机理,并与Aln(n=3~20)团簇与水分子反应机理进行了比较,发现纳米铝团簇与水分子反应机理在不同初始水分子数和温度下存在明显差异.
铝团簇;分子动力学;ReaxFF;氧化
在工业生产中铝是应用最广泛的金属之一,生产过程中极易产生粉尘云,近年来已经发生多起铝粉爆炸事故. 在国防工业中,铝粉作为添加剂广泛应用于含能材料中. 由于纳米铝颗粒具有大比表面积、低活化能和高活性等特点,因此含有纳米铝颗粒的铝粉更容易发生爆炸事故[1],作为添加剂纳米铝粉逐渐取代普通铝粉[2]. 因此,研究纳米铝颗粒的氧化特性具有十分重要的意义,国内外学者对其进行了大量研究[2-3].
一些学者通过实验、理论和数值计算[2-4]对铝团簇与氧气的相互作用进行了研究. 由于实验条件限制,数值模拟在铝团簇氧化过程研究中起到重要作用. Vashishta[5-6]采用ab initio量子动力学对纳米铝团簇与水反应进行了研究,发现产生H2机理:首先,H2O中的O与铝团簇表面具有Lewis酸特性的Al结合,使周围Al的Lewis酸特性提高,而在附近的Lewis碱性位置上通过质子传递产生一个H. 即使H2O自身很难离解,Grotthuss机理[5,7]大幅降低了离解活化能. 最终,铝团簇表面结合H2O中的H和Al-H建中的H结合产生H2,这比水离解产生H2要容易得多. 由于产生OH-具有很强的Lewis碱特性,通过Grotthuss机理反应生成一个H2O,所以一个Lewis酸碱对在酸性条件下可以产生多个H2.
由于ab initio计算量大且计算模型较小,而ReaxFF分子动力学相对量子力学计算量小,能够计算数百万原子构成的体系. ReaxFF已经广泛应用于氧化、燃烧催化等,ReaxFF反应力场被用于对Al—O—H系统[8]研究. 本文采用ReaxFF反应力场对纳米铝团簇在氧气和气相水环境中的初始氧化过程进行了研究,揭示了纳米铝团簇在不同氧化剂初始氧化过程中的氧化规律及反应机理,为防止铝粉爆炸提供了理论依据.
1 计算方法及细节
为模拟纳米铝团簇在氧气和气相水环境中的初始氧化过程,纳米铝团簇分别至于氧气和气相水环境中. 建立一个4 nm×4 nm×4 nm的fcc晶体,晶格常数为0.404 1 nm(T=300 K时). 然后,从剪切出直径为2 nm铝团簇(含249个Al)随后进行退火处理,从而消除边界效应. 把退火后的铝团簇置于10 nm×10 nm×10 nm的计算盒子中心,500个O2随机分布在盒子里,见图1(a). 在铝-水系统中,铝颗粒置于4 nm×4 nm×4 nm的盒子中心,然后把H2O随机分布,初始H2O数分别为50,100,200,500,图1(b)为含200H2O系统示意图. 考虑到工业中产生铝粉尘的环境以及铝粉作为添加剂燃烧环境,模拟温度为600,1 000,1 500,2 000 K. 时间步长设为0.1 fs,Al-O2和Al-H2O系统的计算时间分别为1 000 ps和250 ps.
在300 K条件下用uniform速度分布器生成系统中所有原子的初始速度. 然后对系统进行优化以获得最小能量结构,采用NVT系综对系统进行急剧加温,用Nose-Hoover恒温器控制温度. 在计算过程中每一步都对原子速度进行调节,从而消除了迭代计算误差产生的累积效应. 通过耦合参数控制系统与热浴的温度耦合程度,在Al-O2系统中耦合参数时间步长分别为10 fs和0.1 fs,而Al-H2O系统中耦合参数和时间步长为10 fs和0.1 fs(T=2 000 K时为2.5 fs和0.05 fs). 边界条件为周期性边界条件. 键级设为0.1,通过它确定原子间是否相连,当任意原子对的键级大于0.1时,则认为新的化学键形成,生成新分子.
2 结果与分析
2.1 铝颗粒在氧气环境中的初始氧化过程
图2为Al-O2系统的势能随时间的变化情况. 在初始阶段,系统吸热使势能有少许增加,铝颗粒与吸附O2发生化学反应,导致系统势能逐渐减小. 在600和1 000 K时,系统势能几乎呈线性减小,而在2 000 K,初始阶段(0~200 ps)势能变化平缓,直到约200 ps时势能急剧减小此时发生了剧烈的化学反应,900 ps后趋于平稳. 图4是铝团簇和O2的主要反应路径[9],约200 ps时,系统中出现O导致反应速度显著增大. 图4可知O在反应过程中起到重要作用,它影响整体反应速率. O的出现可能是系统势能急剧减少的原因.
图3展示了不同温度下系统中物种数与时间的关系. 初始时只有Al和O2两种物质,在600 K时O2被吸附到团簇表面,整个模拟过程只有很少铝原子脱离颗粒. 在1 500 K时,在前500 ps物种数逐渐增大,最终维持在15左右. 当温度为2 000 K时,在0~70 ps内,物种数和势能小幅增大,约70 ps时势能达到最大;然后物种数快速增多,在约300 ps时达到最大值(35),随后快速回落;700 ps时物种数基本稳定,此时出现团聚现象,团簇表面形成了氧化薄膜阻止了铝和氧气的进一步反应. 在600 K和1 000 K时,物种主要是铝颗粒团簇和剩余氧分子;在温度为1 500 K和2 000 K时,生成大量铝的氧化物与铝颗粒分离,这可能是实验中观察纳米铝颗粒与氧气反应出现溅射现象[10]的原因.
为了研究温度对纳米铝颗粒在氧气环境中氧化过程的影响,详细地研究了反应产物情况. 图5展示了不同温度条件下Al-O2系统主要产物的演化情况. 由图5可知,主要产物为Al2O3、AlO2、AlO4、AlO、Al和Al2O2等. 其中,仅当T=2 000 K时,系统中出现了单个Al原子. Lynch在实验中发现纳米铝颗粒在空气氧化,仅当T>1 500 K时才会出现Al原子,这与模拟的结果一致. 单独的Al原子先增大后减少,约250 ps达到最大,在400 ps后趋于稳定直至600 ps时完全消失,这与O2在400 ps后消耗趋于稳定相符. 对T=1 500 K和T=2 000 K进行对比分析,AlO2是早期主要中间产物,从图中可以看出不同温度下AlO2的分布具有相似性,均是先增加后减少的过程. 在2 000 K时,AlO2峰值要比1 500 K大得多,达到峰值后减少的速度也要快得多,由此可见,温度对纳米铝颗粒具有显著的影响.
图6为不同温度下消耗氧气分子数随时间变化的曲线. 在反应初期,消耗氧气速度较快,然后,在600,1 000和1 500 K时,消耗氧气分子呈线性关系. 这是以为反应速率是由团簇表面氧气扩散速率决定的. 在600 K时,在950 ps后,消耗氧分子数基本保持不变,这是因为团簇表面形成氧化膜阻止铝和氧气进一步反应. 在2 000 K时,O2在前期逐渐消耗,约400 ps O2消耗趋于平缓,到800 ps时O2消耗数量出现小幅下降.
2.2 铝颗粒在水蒸气中氧化2.2.1 系统势能演化
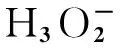
2.2.2 物种演化分布
图8为Al-H2O系统在不同温度条件下总物种随时间的变化曲线.
由图8可知,温度和水分子数对总物种数演化影响十分显著. 从图8(a)可知,有限模拟时间内,当温度为600~1 500 K时没有新物种生成,当2 000 K时有少量新物种生成;在图8(b)、(c)和(d)中,物种数演化趋势相似,初始H2O越多,在模拟中物种数越多. 在同一系统中,温度对物种数影响明显,从图7(d)可知,温度越高物种数增长的越快,在高温时(2 000 K)总物种数先快速增多达到最大值后快速回落,然后保持不变;低温时(600 K)总物种数逐渐增长. 不同Al-H2O系统在2 000 K时总物种数的分布,总物种数先增大后减少,初始水H2O越多,物种数起初增长得越快. 当初始H2O数较少时,温度对系统的反应过程影响不明显;当初始H2O数较多时,温度对系统的反应过程有显著的影响.
2.2.3 水分子数量对反应过程的影响
图9为不同 Al-H2O系统在不同温度下剩余H2O的数量与时间的关系. 可以看出,初始阶段水分子数急剧减小,在50 ps吸附过程结束,初始H2O数对反应影响明显. 在Al-50H2O和Al-100H2O系统中,H2O数急剧减少,约60 ps时剩余水分子数趋于稳定,温度越低团簇吸附和消耗水分子过程越快;而在Al-200H2O和Al-500H2O系统中,温度越低铝团簇吸附和消耗水分子的过程越慢. 这可能是由于不同H2O数量导致了反应机制变化,H2O较少时主要是吸附过程,温度越低吸附越快;H2O较多时发生化学反应,此时温度越高化学反应越剧烈,H2O消耗越快. 对比图9可知,Al-50H2O和Al-100H2O系统中,在0~50 ps基本没有H3O+和H2生成,而在Al-200H2O和Al-500H2O系统中,初始阶段大量H3O+生成. 在Al-50H2O系统中,在600和1 000 K时,分别在约60 ps和110 ps铝团簇吸附了所有的H2O. 而在Al-500H2O系统中,在1 500 K时,剩余水分子数达到最小值后逐渐增多;而在1 000 K时,水分子几乎被完全吸收和消耗.
图10为不同Al-H2O系统分别在1 500 K和2 000 K时被吸附和消耗的水分子数与时间的关系. 在相同温度时,初始水分子数越多水分子被吸收和消耗得越快,然后到达稳态阶段,这是因为铝颗粒表面覆盖完全,阻碍了进一步反应;当温度为1 500 K时,在Al-200H2O和Al-500H2O系统中,当吸收和消耗的水分子数存在一个最大值,达到最大值后吸收和消耗的水分子数存在一个小幅回落过程.
2.2.4 反应产物及反应机理
通过分析Al-H2O系统的反应产物可以发现,在Al-50H2O、Al-100H2O和Al-200H2O系统中,仅当温度为2 000 K时,产物中有H2生成;而在Al-500H2O系统中,当600 K时没有H2生成,其他温度均有H2生成. H2逐渐积累,在2 000 K时,系统中初始H2O分子数越多,则越早产生H2而且H2的数量也越多.
对于Al-50H2O系统,仅在2 000 K时,有极少的H3O+生成;然而,在600 K时,只有Al-500H2O系统中才出现H3O+. 在Al-100H2O、Al-200H2O和Al-500H2O系统中,在1 000、1 500和2 000 K时均有H3O+生成. 从图11可知,水分子数越多,H3O+越多反应越剧烈,且整体趋势相似,H3O+数量先增大后减小. 这是因为团簇表面铝原子和H2O通过“特殊”机理快速生成H3O+,随着反应进行,团簇表面活性铝和O不断结合,阻碍了与H2O结合从而减少了H3O+生成;而H3O+和团簇表面Al-H反应生成H2,使H3O+不断消耗.

图13为Al-500H2O系统在不同温度下H2和H3O+的演化曲线. 温度越高则越容易生成H2,在600 K时,模拟时间内未生成H2. 在不同温度时H3O+演化过程相似,先增大后减少. 对比图13(b)和图9(d)可以发现:当T=2 000 K时,40 ps时达到最大值,此时水分子数达到最小值且此后趋于稳定.
3 结 论
使用ReaxFF反应力场分别对Al-O2系统和Al-H2O系统的初始氧化过程进行了模拟. 结果表明,Al-O2系统在高温时条件下,大量铝的氧化物和单个Al原子脱离铝团簇. 铝团簇与氧气反应过程中是否出现单个Al原子是由温度决定的,且存在一个温度阈值,当温度低于1 500 K时不会出现单个Al原子,在2 000 K时则出现单个Al原子.
在直径为2 nm的铝团簇和水分子组成的系统中,铝团簇表面的水分子对铝和水反应具有促进作用,从而降低了铝和水反应的能量壁垒.
系统初始水分子数以对直径为2 nm铝团簇初始反应过程的影响十分显著. 存在两个明显不同的反应机制,初始水分子数为50的Al-H2O系统在模拟时间内未发生化学反应,水分子全部被铝团簇吸附在其表面,而当水分子数增多时,铝团簇和水分子进行反应,初始水分子数越多,反应越快.
在Al-O2系统中,反应中间产物O出现导致反应速率大幅提高;而在Al-H2O系统的反应过程中,H3O+作为重要的中间产物,通过和铝团簇表面的H2O和OH反应对整个反应过程产生重要的影响. 在Al-O2系统中,温度对系统反应过程具有显著的影响;而在Al-H2O系统反应过程中,初始水分子数较少时,温度对系统影响不显著,而当初始水分子较多时,温度对系统反应过程具有显著的影响.
[1] Li Q, Lin B, Li W, et al. Explosion characteristics of nano-aluminum powder-air mixtures in 20 L spherical vessels[J]. Powder Technology, 2011,212(2):303-309.
[2] Pantoya M L, Granier J J. Combustion behavior of highly energetic thermites: nano versus micron composites[J]. Propellants, Explosives, Pyrotechnics, 2005,30(1):53-62.
[3] Eisenreich N, Fietzek H, Del Mar J L M, et al. On the mechanism of low temperature oxidation for aluminum particles down to the nano-scale[J]. Propellants, Explosives, Pyrotechnics, 2004,29(3):137-145.
[5] Fuyuki S, Ohmura S, Kalia R K, et al. Molecular dynamics simulations of rapid hydrogen production from water using aluminum clusters as catalyzers[J]. Physical Review Letters, 2010,104(12):126102.
[6] Shimojo S A, Kalia F A, Kunaseth R K A, et al. Reaction of aluminum clusters with water[J]. Journal of Chemical Physics, 2011,134(24):244702.
[7] Marx D, Tuckerman M E, Hutter J, et al. The nature of the hydrated excess proton in water[J]. Letters to Nature, 1999,397:601-604.
[8] Russo M F, Li R, Mench M, et al. Molecular dynamic simulation of aluminum-water reactions using the ReaxFF reactive force field[J]. International Journal of Hydrogen Energy, 2011,36(10):5828-5835.
[9] Huang Y, Risha G A, Yang V, et al. Analysis of nano-aluminum particle dust cloud combustion in different oxidizer environments[C]∥Proceedings of the 43rd Aerospace Sciences Meeting and Exhibit. Reno, Nevada: [s.n.], 2005:2005-2738.
[10] 炎正馨.激波诱导下纳米铝粉与微米铝粉的爆炸特征对比研究[J].物理学报,2011,60(7):526-530.
Yan Zhengxin. Compare study on the explosion characteristics of nano-aluminum and micro-aluminum[J]. Acta Physica Sinica, 2011,60(7):526-530. (in Chinese)
(责任编辑:刘雨)
Molecular Dynamics Simulation of Initial Oxidation Process of Nano-Aluminum Clusters
YANG Zhen, HE Yuan-hang
(State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology, Beijing 100081, China)
The oxidation characteristics and mechanism of aluminum cluster in different oxidizers were studied. ReaxFF force field was used to simulate the initial oxidation process of Al-O2system and Al-H2O system respectively. Results show that in Al-O2system, the temperature of environment decides on whether there exists Al atom around aluminum cluster during initial oxidation. Moreover, there exists a temperature threshold value, below that the Al atom does not exist. For Al-H2O system, the influences of the number of initial water molecules and the temperature were studied. Studies show that water tends to promote the dissociation of water molecules absorbed by aluminum cluster. Reaction mechanism of aluminum cluster(2 nm) and water molecule were also discussed and compared with that of Aln(n=3~20)cluster and water. The results show that there exists evidence difference in the reaction mechanism when the number of initial water molecule and temperature are distinct.
aluminum cluster; molecular dynamics; ReaxFF; oxidation
2015-02-13
杨镇(1989—),男,博士生,E-mail:yzhen278@163.com.
O 642; O 643
A
1001-0645(2016)08-0868-08
10.15918/j.tbit1001-0645.2016.08.017
