强直性肌营养不良1型患者的眼部症状分析
2016-11-22阎雪晶欧阳嶷何志义崔小雪朱华倩史铭宇王欣玲
阎雪晶,欧阳嶷,何志义,崔小雪,朱华倩,史铭宇,王欣玲
(中国医科大学1.附属第一医院神经内科,沈阳 110001;2.附属第四医院眼科,沈阳 110005)
·论著·
强直性肌营养不良1型患者的眼部症状分析
阎雪晶1,欧阳嶷1,何志义1,崔小雪1,朱华倩1,史铭宇2,王欣玲2
(中国医科大学1.附属第一医院神经内科,沈阳 110001;2.附属第四医院眼科,沈阳 110005)
目的 分析5个家系强直性肌营养不良1型(DM1)患者眼部表现,总结其临床特点,为早期诊断提供依据。方法 总结5个家系已确诊的患者进行强直性肌营养不良蛋白激酶(DMPK)基因CTG重复次数,归纳患者临床症状及特点。结果 患者均表现为肌无力、肌强直;患者均患白内障,有视物模糊、上睑下垂等表现;肌电图提示肌源性损害,肌强直电位;裂隙灯显微镜及眼科超声生物显微镜提示周边后囊前皮质片状混浊;4例患者DMPK基因CTG重复>50次,2例DMPK基因CTG重复50次。结论 眼部症状是DM1的重要表现之一,其严重程度与CTG重复序列数之间并无明显关联。
强直性肌营养不良1型;眼部症状;白内障;基因检测
强直性肌营养不良1型(myotonic dystrophy type1,DM1)是一种逐渐进展的多器官、多组织受累的常染色体显性遗传疾病,主要表现为肌强直、肌萎缩、心肌传导阻滞、白内障、内分泌紊乱等。DM1发病率在不同地区波动于1∶8 000(加拿大)至1∶20 000(亚非地区)之间,是成人最常见的肌萎缩疾病[1]。DM1是由于位于19q13.3的强直性肌营养不良蛋白激酶(myotonic dystrophy protein kinase,DMPK)的CTG序列异常扩增引起。正常人的CTG序列扩增数目为5~37;当CTG序列扩增数目为38~49时为前突变期,携带者本人无异常表现,但其后代此序列扩增数目较常人增多且可有部分此疾病的临床表现;当CTG序列扩增数目≥50时为全突变期,表现出明显的临床症状,且疾病的严重程度与扩增序列的多少有一定关系[2]。目前DM1主要通过基因检测来确诊。然而在疾病早期,白内障可能是该疾病的唯一临床表现[3],因此,为了早期发现并诊断DM1,本研究对5个家系6例经基因检测确诊的DM1患者进行详细的眼科查体及仪器检查,总结分析DM1患者眼部症状特点,从而为早期诊断提供依据。
1 材料与方法
收集2013年7月至2014年12月中国医科大学附属第一医院神经内科门诊及病房的5个家系6例DM1患者的临床资料,年龄34~53岁,平均45岁,病程2~20年,平均11年。对上述患者进行神经内科学及眼科学相关检查,并抽取受检者外周血(2 mL)进行基因分析。全部研究对象均签署了知情同意书。
2 结果
2.1 家系及临床表现分析
患者家系图见图1。所有患者均慢性起病,主要症状为肌无力、肌萎缩,肌无力均为左右对称,且远端、近端肌力无明显差别,肌萎缩以四肢肌肉为主。所有患者体格检查均可见肌病面容,构音不良;叩击舌肌有肌球形成。此外,家系2先证者Ⅱ3、家系3Ⅲ1有周围性面瘫。肌电图(electromyography,EMG)显示肌源性损害,部分患者可见肌强直电位(表1)。除家系4先证者、家系5先证者CTG扩增序列为50,其余均>50,见表2。

图1 家系图Fig.1 Family tree
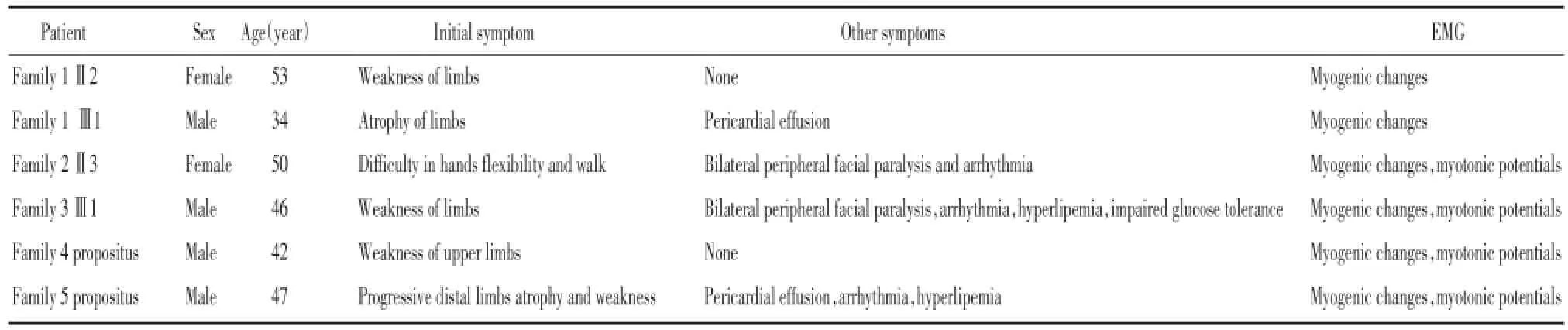
表1 DM1患者临床表现及EMG结果Tab.1 Clinical manifestations and findings of DM1 patients
眼部症状及检查显示,除1例患者(家系1Ⅲ1)有屈光异常外,所有患者都表现为晶状体后囊前皮质混浊(图2),并导致视力不同程度下降,未发现其他视功能及眼部形态异常改变。所有患者均有上睑下垂,另外,2例患者(家系5先证者、家系2Ⅱ3)有复视,1例患者(家系5先证者)眼位异常(图3、表2)。
3 讨论
DM1在眼部的特征性表现为进行性上睑下垂、眼外肌麻痹、特征性白内障、视网膜色素沉着、睫状体异常、眼压降低等[4,5]。本研究5个家系中6例确诊患者均有上睑下垂症状,其中2例还伴有周围性面瘫表现,累及肌肉为上睑提肌及眼轮匝肌,受累神经为动眼神经及面神经,而面神经麻痹可导致睑板腺功能障碍,从而导致眼部异物感、灼烧感、视物疲劳、流泪、眼红、视物波动等[6,7],与患者描述症状相吻合。上述症状出现的时间大多同时或晚于肌萎缩、肌无力症状出现的时间,体现了强直性肌营养不良不仅累积四肢骨骼肌还同时累积面部肌肉的特点。而家系2Ⅱ3患者出现复视,说明眼外肌受累。它的病理表现与骨骼肌相似,为坏死肌纤维的聚集即破红肌纤维的形成,而当大部分肌纤维被结缔组织替代时提示进入疾病晚期[8,9]。

表2 患者眼部表现及基因检测结果Tab.2 Ocular manifestations and gene test results of patients
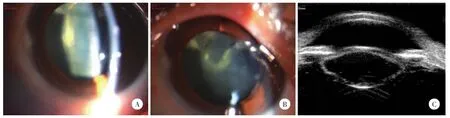
图2 家系1Ⅱ2患者裂隙灯显微镜及眼科超声生物显微镜(频率25 MHz)检测结果Fig.2 Family1Ⅱ2 results of slit lamp microscope and ultrasonic biological microscope(Frequency=25 MHz)

图3 家系5先证者眼部表现Fig.3 Family 5 propositus ocular manifestations
眼部并发症中最具特征、最常见的为白内障,其特征性表现为后极部彩虹样点片状混浊[10],它可能与晶状体上皮细胞的缺乏有关[11]。特征性白内障作为强直性肌营养不良一个主要的甚至唯一的临床特征为早期诊断提供重要临床依据[12]。有报道表明[13]:白内障极早出现的病例高达90%,但引起视力明显减退者占少数,因此早期诊断常被忽略。本研究5个家系6例患者均患有白内障,表现为后囊前(后极部或周边部)皮质灰白混浊,大部分患者眼部症状出现晚于强直性肌营养不良的主要临床症状,而1例患者(家系3Ⅱ2)仅有眼部表现而无肌病改变。6例患者中家系1Ⅱ2患者的白内障程度最重,家系1Ⅲ1患者的白内障程度最轻,而家系4先证者已行双眼白内障手术,故考虑其白内障程度较为严重。而从CTG扩增序列来看,只有家系4先证者及家系5先证者CTG为50,其余均>50。从中可以看出患者白内障的严重程度与CTG扩增数目的多少并无相关性。
从发病年龄来看,患者都为中年起病,3例患者因明显的视力下降、视物成双等症状就诊,其余患者无明显临床症状,与许波等[14]报告的家系相似。从临床表现来看,其严重程度不尽相同:有的患者仅有轻微视力改变(家系3Ⅲ1),而有的患者因视力障碍已行白内障手术(家系4先证者),但2者肌无力表现相近,因此认为白内障的严重程度与疾病全身表现的严重程度并不一致,这与Simon等[10]的观点一致。从白内障症状进展情况来看,患者肌无力、肌萎缩症状出现加重时,视物不清等症状无明显变化,提示白内障的进展与肌无力、肌萎缩的进展并无明显联系。从预后来看,若患者视力不影响正常生活,则可保守治疗,定期观察;若患者视力影响生活质量,则可行手术治疗,但需定期观察,注意后囊混浊和前囊挛缩的并发症[15]。
另外本研究结果显示家系5先证者有青光眼病史。该患青光眼出现晚于肌无力、肌萎缩症状,且并未随DM1的疾病进展而加重。目前尚无关于DM1与青光眼之间存在联系的报道,需进一步动态观察其青光眼进展情况,并在今后的研究中收集更多的病例,明确青光眼是否为与DM1相关的眼部症状。
针对患者视力改变,通过裂隙灯显微镜(需要散瞳检查),尤其是眼科超声生物显微镜检查能够明确白内障的特征性改变,即晶状体后囊前皮质片状混浊,从而将此结果作为诊断依据之一。DM1的确诊方法为基因诊断,不能仅凭肌电图及裂隙灯下特征性白内障改变而确诊DM1,既往很多误诊病例[16]需要引起临床神经科医生和眼科医生的注意。此外,对于中老年DM1患者,需注意与老年性白内障相鉴别,以免误诊。有研究[17]表明患者视力改变可能与视网膜前膜有关,并不一定都为白内障所致,因此临床工作者需谨慎鉴别。
[1]Emery AEH.Population frequencies of inherited neuromuscular diseases-a world survey[J].Neuromuscular Disorders Nmd,1991,1(1):19-29.
[2]Gonzalez I,Ohsawa N,Singer RH,et al.New nomenclature and DNA testing guidelines for myotonic dystrophy type 1(DM1)[J]. Neurology,2000,54(6):1218-1221.
[3]Papageorgiou E,Bock SW,Schiefer U.Myotonic dystrophy curschmann-steinert[J].Klinische Monatsblätter Für Augenheilkunde,2007,224(1):70-75.
[4]何苗.强直性肌营养不良症20例临床分析[J].中国现代药物应用,2015,9(3):160-161.
[5]Mayer C,Cordeiro SA,Khoramnia R.Diagonose:bilaterale cataracta subcapsularis posterior bei myotoner dystrophie curschmann-steinert[J].Der Ophthalmol,2011,108(10):976-980.
[6]Call CB,Wise RJ,Hansen MR,et al.In vivo examination of meibomian gland morphology in patients with facial nerve palsy using infrared meibography[J].Ophth Plast Reconstr Surg,2012,28(6):396-400.
[7]Shah CT,Blount AL,Nguyen EV,et al.Cranial nerve seven palsy and its influence on meibomian gland function[J].Ophth Plast Reconstr Surg,2012,28(3):166-168.
[8]Isashiki Y,Kawabata E,Ohba N,et al.Mitochondrial abnormalities in extraocular muscles in myotonic dystrophy[J].Neuro Ophthalmol,2009,9(2):115-122.
[9]Kuwabara T,Lessell S.Electron microscopic study of extraocular muscles in myotonic dystrophy[J].Am J Ophthalmol,1976,82(2):303-309.
[10]Simon KA.Diabetes and lens changes in myotonic dystrophy[J]. Arch Ophthalmol,1962,67(1):312-315.
[11]Abe T,Sato M,Kuboki J,et al.Lens epithelial changes and mutated gene expression in patients with myotonic dystrophy[J].Br J Ophthalmol,1999,83(4):452-457.
[12]Su JJ,Chen X,Dong ZO.Characteristics of cataract in 26 patients with myotonic dystrophy from 3 pedigrees[J].J Clin Rehab Tis Engin Res,2007,11(51):10413-10415.
[13]龙秀英,刘鸣.强直性肌营养不良97例临床资料分析[J].中国循证医学杂志,2004,4(3):194-197.
[14]许波,唐北沙,张玉虎,等.强直性肌营养不良一家系报告[J].中华神经科杂志,2003,36(6):483.
[15]Garrott HM,Walland MJ,O'Day J.Recurrent posterior capsular opacification and capsulorhexis contracture after cataract surgery in myotonic dystrophy[J].Clin Exper Ophthalmol,2005,32(6):653-655.
[16]Barnes PR,Hilton-Jones D,Norbury G,et al.Incorrect diagnosis of myotonic dystrophy and its potential consequences revealed by subsequent direct genetic analysis[J].J Neurol Neurosurg Psychiatry,1994,57(5):662.
[17]Kersten HM,Roxburgh RH,Nicholas C,et al.Epiretinal membrane:a treatable cause of visual disability in myotonic dystrophy type 1[J].J Neurol,2014,261(1):37-44.
(编辑 武玉欣)
Ocular Symptoms Analysisof Myotonic Dystrophy Type1 Patients
YANXue-jing1,OUYANGYi1,HE Zhi-yi1,CUIXiao-xue1,ZHUHua-qian1,SHIMing-yu2,WANG Xin-ling2
(1.Department of Neurology,The First Hospital,China Medical University,Shenyang 110001,China;2.Department of Ophthalmology,The Forth Hospital,China MedicalUniversity,Shenyang 110005,China)
Objective To summarize the clinical features of ocular symptoms analysis in 5 families of myotonic dystrophy type 1(DM1)patients,so as to increase the reorganization of DM1 for clinician and provide the basis for early diagnosis.Methods Five genetically diagnosed DM1 subjects were enrolled in the study,and the number of trinucleotide CTG in the region of myotonic dystrophy protein kinase(DMPK)and the research of clinical symptoms were determined,then the clinical manifestation were summarized.Results All the patients were manifested with myasthenia and myotonia,cataract,blurred vision and ptosis;resultsofelectromyography showed muscle damage and myotonic potential;results ofslit-lamp microscope and ophthalmic ultrasound biomicroscope showed patchy opacities of the anterior cortex of the posterior capsule;the number of CTG fragment of 4 patients exceeded 50,and the other 2 were 50.Conclusion Ocular symptom is one of the most important manifestations of DM1;there is no clearcorrelation between the severity ofDM1 and numberofsequences CTG.
myotonic dystrophy type 1;ocular symptom;cataract;gene test
R746.2
A
0258-4646(2016)02-0097-04
10.12007/j.issn.0258-4646.2016.02.001
国家自然科学基金青年基金(81100879)
阎雪晶(1991-),女,硕士研究生.
欧阳嶷,E-mail:oyyxmx@hotmail.com
2015-11-02
网络出版时间:
