不可逆蛋白激酶抑制剂研究进展
2016-11-17郑培根郑程隆
郑培根,郑程隆
(青岛科技大学,山东青岛 266042)
不可逆蛋白激酶抑制剂研究进展
郑培根,郑程隆
(青岛科技大学,山东青岛 266042)
蛋白激酶是一个由多种高度保守酶组成的酶系,主要负责将ATP的Y-磷酸基团转移给酪氨酸、丝氨酸、苏氨酸等多种氨基酸,这些氨基酸在细胞中起到信号传导的作用。迄今为止,多数上市的激酶抑制剂是ATP竞争的非共价抑制剂,但是近来不可逆蛋白抑制因其可以和半胱氨酸或其他亲核残基在ATP口袋处结合形成共价键而备受科研人员的青睐。通过讲述不可逆蛋白激酶抑制剂的研究进展,探讨新型共价抑制剂的高效发展战略。
蛋白激酶;共价键抑制剂;不可逆性
激酶是一个大的基因家族,功能是催化ATP上的Y-磷酸盐转移到特定的靶标分子。蛋白质磷酸化可以诱导产生多种影响,包括酶的活性、构象、稳定性等[1]。在所有激酶中,蛋白激酶家族是一个很有吸引力的药物靶点。目前,在所有药理学靶点中,有超过25%是蛋白激酶,而在抗肿瘤领域更是有超过75%的靶点是蛋白激酶[2-3]。目前,上市的大多数蛋白激酶抑制剂均为可逆性的ATP竞争抑制剂,其通过识别特定的ATP结合口袋,从而具有靶向性、选择性等特点[4]。这类抑制剂能够和ATP结合口袋处的腺嘌呤结合,并可延伸到周围。抑制剂的结合经常会引起显著性的结构重排,进而影响生物的新陈代新[5]。
不可逆激酶抑制剂的发现相对较晚,但是其应用领域不断扩大。以下2个方面的发展,对不可逆激酶抑制剂的开发有重要影响。首先,在过去的十几年里,小分子抑制剂在医学领域取得飞速发展。这些激酶抑制剂获得美国药物食品监督管理局
(FDA)批准,并且用于肿瘤治疗。但是这些蛋白激酶抑制剂都是使用传统方法识别、开发的,即这些可逆激酶抑制剂占据了ATP结合口袋中激酶的催化结构域。第二个重要的发展是共价结合[6-7],这主要源于共价键药物选择性和安全性方面的全新认识。历史经验告诉药物研发者,要避免使用亲电的小分子化合物。因为小分子亲电基团的选择性会非常混乱,或者即使选择性很好,在后期的临床试验中也会表现出很高的毒性。其结果是不可逆抑制剂或者共价键抑制剂成为毒性试剂,并且开发的也越来越少。但是随着临床上不可逆抑制剂有更好的疗效和安全性例子的增多,对不可逆抑制剂的质疑才不断减少。概括来说,不可逆抑制剂能否成功取决于共价键是否只局限于靶向的蛋白激酶,即包括疗效和毒性2个方面的内容。
1 常见不可逆激酶抑制剂的种类
1.1 Bruton's酪氨酸激酶(BTK)抑制剂
Bruton's酪氨酸激酶是非受体酪氨酸激酶中Tec家族的一员,也是一种在B淋巴细胞的生长、增殖、分化中起着重要作用的酪氨酸激酶,因此它是B细胞淋巴瘤的关键靶标酶。由于在免疫应答中涉及到B细胞,因而BTK抑制剂也可以应用于自身免疫性病症。BTK在铰链结合区拥有1个非催化性的Cys481,即非常接近于ATP结合位点的抑制剂。BTK抑制剂表现出良好的临床疗效和安全性,该抑制剂的成功开发对Pharmacyclics'公司的Imbruvica(拉铁尼伯)用于治疗外套细胞淋巴瘤(MCL) 的批准有很大推进作用,该药物的口服生物利用度较高[8]。Pharmacyclics'公司目前正在与Jansen医药公司合作,研究Imbruvica(拉铁尼伯)在治疗慢性淋巴细胞白血病(CLL)、多发性骨髓瘤和弥漫性大B细胞淋巴瘤上的作用。
常见Bruton's酪氨酸激酶(BTK)抑制剂见图1。
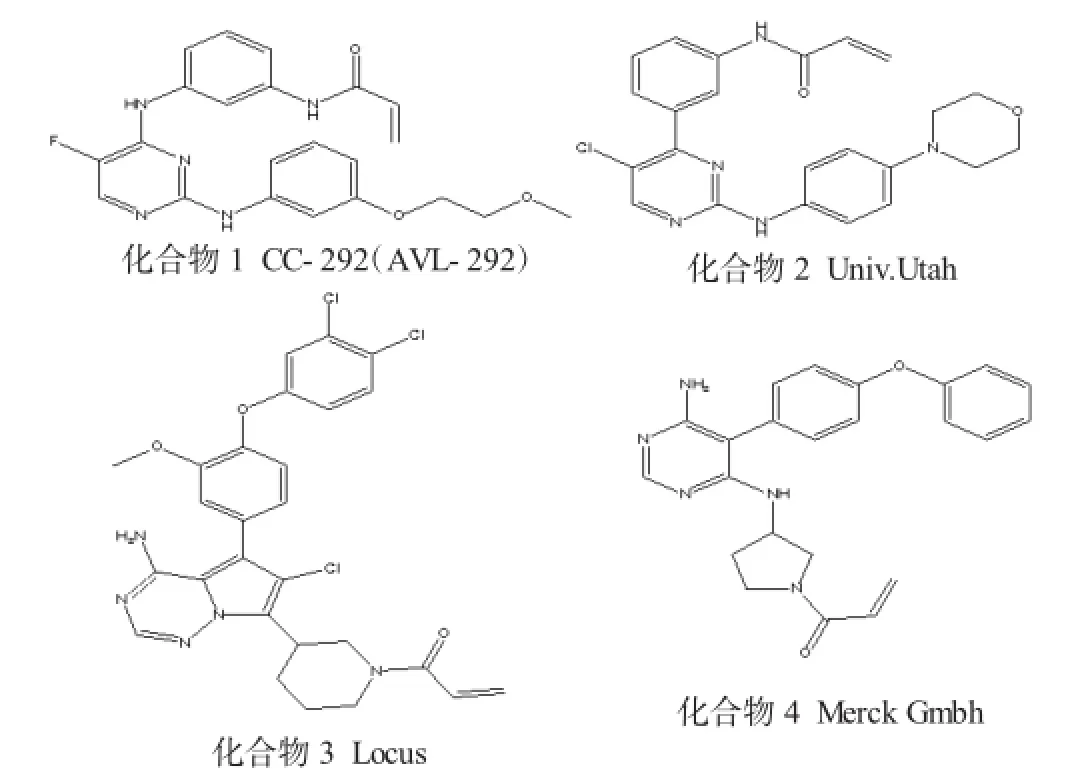
图1 常见Bruton's酪氨酸激酶(BTK)抑制剂
1.2 表皮生长因子受体(EGFR)抑制剂
EGFR激酶是一种细胞表面的酪氨酸激酶,它们在激活细胞信号转导过程起到重要的作用。EGFR含量的上升是许多癌症的病因,包括肺癌、结肠癌和胶质母细胞瘤。许多制药公司研究不可逆EGFR激酶抑制剂已超过15年,正是由于他们的坚持,才使很多第1代不可逆抑制剂提前进入临床试验。转移性非小细胞肺癌(NSCLC)是由于野生型EGFR的过度表达,外显子19的缺失或外显子21 L858R的突变引起。为了制止该疾病的蔓延,2013年美国批准上市了Gilotrif(Afatinib) 并作为一线治疗药物[9]。另外2种与EGFR轮廓类似的抑制剂Neratinib和Dacomitinib[10],正处于III期临床试验阶段。所有这些抑制剂均含有丙烯酰胺基结构,该结构与EGFR亚型中Cys797在铰链结合区有不可逆地相互作用。最近研发的新型EGFR抑制剂焦点都集中在抑制EGFR的双突变T790M/L858R,因为它们可能是大多数癌症患者用EGFR抑制剂治疗的耐药机制[11]。常见表皮生长因子受体(EGFR)抑制剂见图2。
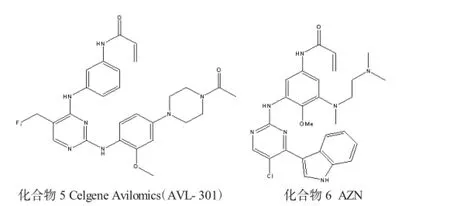
图2 常见表皮生长因子受体(EGFR)抑制剂
1.3 Janus激酶3(JAK3)抑制剂
在胞浆中的JAK3是Janus家族中酪氨酸受体激酶(JAK1,JAK2和TYK2)之一,它们参与细胞因子信号传导过程。具有选择性的JAK3抑制剂能够较好地治疗许多自身免疫适应症,如类风湿性关节炎、炎性肠病和器官移植排斥反应等。此外,JAK3抑制剂还是一个有价值的、选择性更强的药物,最近推出的JAK3抑制剂Xeljanz(Tofacitinib),已获用于治疗类风湿性关节炎[12]的批准。JAK3在其铰链结合区具有独特的、不存在于JAK1,JAK2和TYK2的半胱氨酸残基(Cys909),最近的专利文献也经常开始引用不可逆JAK3抑制剂的例子,因为JAK3抑制剂可以与JAK3 Cys909结合,进而产生选择性。
常见的JAK3抑制剂见图3。
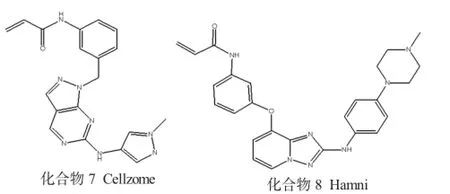
图3 常见的JAK3抑制剂
1.4 磷脂酰肌醇-3-激酶抑制剂
磷脂酰肌醇-3-激酶(PI3Ks)是受体酪氨酸激酶下游的主要稳压器,能够调节许多主要的细胞功能,如细胞生长、分化、运输和凋亡。这些激酶的失调,往往暗示着许多转移性癌症[13]的发生。非催化半胱氨酸(PI3KαCys862)位于PI3Ks的铰链结合区后,不可逆抑制剂正是以此为靶点实现与酶的结合。在该领域,Celgene Avilomics公司一直通过合成含有双环杂环的化合物进行专利申请,如化合物9结构中的噻吩并嘧啶丙烯酰胺。
常见PI3Ks抑制剂见图4。
1.5 其他不可逆的激酶抑制剂
除了前面所讨论的主要激酶靶点外,还有专利记载以其他激酶为靶点的不可逆抑制剂。Avila公司
R提出了一种识别激酶的方法申请,即在与可逆抑制剂的结合位点周围处包含Cys残基的激酶。由已知的可逆性抑制剂与酶的结合得出,只要不可逆抑制剂包含亲电基团便可以与该半胱氨酸形成共价键[14]。化合物14是FAK不可逆抑制剂的一个例子,该抑制剂能靶向与FAKP环Cys427结合。Taiho公司已经公开了一系列含有吡唑并嘧啶的FGFR抑制剂,这些抑制剂的结构与以前讨论过[15]的吡唑并嘧啶BTK抑制剂相似。化合物15是由Barcelona大学研制出来的GSK 3β不可逆性抑制剂[16],关于化合物15的可逆性类似物如何通过改造(与DFG模段附近的Cys199相互作用) 而成为不可逆性抑制剂的描述都在文献[17]中有描述。此类例子还有很多,不再逐一列举。
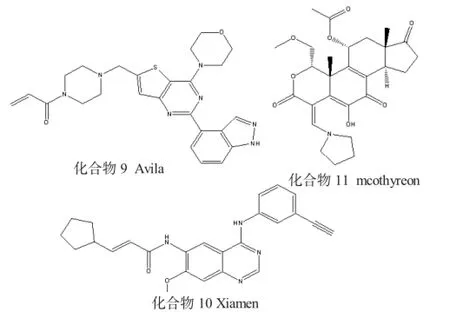
图4 常见PI3Ks抑制剂
其他不可逆的激酶抑制剂见图5。
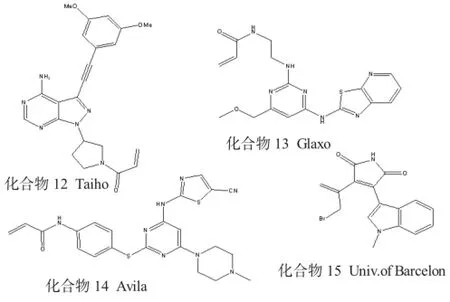
图5 其他不可逆的激酶抑制剂
2 优势与劣势
抑制剂的选择性和安全性一直是发展共价抑制剂治疗肿瘤进程中的重要问题。令人担忧的是,一个不可逆抑制剂会杂乱地在体内标记多个目标,导致细胞毒性或免疫毒性。在本次审查中所列的专利文献表明,许多公认的BTK和EGFR抑制剂对其他酶的抑制能力比对原始酶的抑制能力还强,尤其是那些靶向与激酶中保守的非催化Cys铰链结合区结合的化合物。尽管不可逆激酶抑制剂存在选择性的问题,但是已批准的不可逆激酶抑制剂Afatinib和Ibrutinib显示出良好的安全性[18]。FDA已批准这些药物用于肿瘤适应症,说明具有较温和的不良反应,因此可用于未满足的医疗需求领域。
由于许多不可逆激酶抑制剂对很多的靶点突变体都会产生很强的作用效力(尤其是EGFR抑制剂),所以不可逆抑制剂一般只作为二线抗肿瘤药物[19]。但是最近新公开的不可逆抑制剂也可作为一线药物治疗患者,前提是患者的肿瘤基因分型显示其突变体的表达对已知的标准疗法产生耐药性[20]。因此,不可逆激酶抑制剂可以为这些患者提供更大的初始效力和潜在完善的安全性。
总之,不可逆蛋白激酶抑制剂最显著的优点是对靶标的活性强,一旦药物分子经过代谢到达靶标组织与靶蛋白结合后,抑制活性将不再依赖于血药溶度(AUC)、半衰期(t1/2)等药动数据[9]。因为不可逆小分子的结合,将使被结合的靶蛋白永久失去生物功能,而机体只能通过自身新陈代谢重新生成靶蛋白来恢复本身的功能。这样又会带来另一个问题,即毒性问题。因为一旦药物分子错误地结合了非靶标蛋白,将会影响正常的生理功能。
3 发展策略
早期报道的“人类灵感”共价抑制剂的构建是通过修改已知的非共价抑制剂,即在适当的位置上亲电试剂接近半胱氨酸残基,进而两者发生反应。随着科研人员不断对新激酶配体的发现,可用的复杂结构配体数目也随之增加,这一现象就为这种方法提供了起始点。但是,该方法的瓶颈是寻找模板,使之能在微摩尔浓度范围内与非共价键结合,并能在等量、位置确定的半胱氨酸激酶中展现出选择性。如果共价键的形成是成功的,那么相对于没有适当位置的半胱氨酸残基所有激酶来说,该共价键是有选择性的。另一项挑战是获得脚手架,它可以呈现一个合适的“平台”,该平台安装了带电基团相对亲核半胱氨酸的正确轨迹。ATP位点的识别元素和亲电子之间的活性越少,说明单个半胱氨酸残基作为目标的可能性就越高。在这种情况下,理想的化合物是最初与激酶的结合模式,所处的几何空间能够快速成键形成亲电子的化合物[21]。
尽管有关报道称共价抑制剂是合成的,但是也有许多天然产物通过共价修饰激酶ATP结合位点处的半胱氨酸残基形成共价抑制剂。其中,有一类共价激酶抑制剂最明显的表征是结构中包含二羟基苯甲酸酯类化合物(RALs),Hypothemycin是含有该结构的著名药物之一。Hypothemycin最初是根据其抗真菌活性被应用于临床,而随后的调查证明它是一个共价蛋白激酶抑制剂。共价键是通过结构中顺式烯酮与半胱氨酸残基反应而形成的[22]。
4 展望
许多激酶对多种恶性肿瘤的发作有关,并会起到关键的调节作用,从而使该蛋白家族成为一个有吸引力的药物靶标。目前,研究以激酶为靶点的药物难点在于发现和确定更多的潜在激酶抑制剂,并合成出选择性更强、效果更佳、安全范围更广的药物。与可逆性激酶抑制剂相比,现阶段酶领域的发展更适合具有明确潜在优势的不可逆性激酶的发展。首先,细胞内ATP含量很高,非ATP竞争是一个主要的上涨空间。第二,人类激酶组在ATP结合口袋内或附近有一个特定半胱氨酸,这不仅增强不可逆激酶抑制剂的效力也实现了选择性。第三,许多激酶能够有效地与不可逆抑制剂结合形成共价键,使得该类酶的更新率降低,进而导致作用于此蛋白家族的药物不必频繁给药。这些发展变化,使研究人员对不可逆激酶抑制剂作为有效和安全的治疗方法越来越有信心。见证已经用于治疗危及生命的癌症药物Afatinib和Ibrutinib,能否可以扩展到治疗慢性疾病(类风湿性关节炎)是非常有趣的。在不久的将来,科研人员一定会进一步完善不可逆激酶抑制剂的多种功效,将其毒副作用降低到最小,期待这类药物的早日问世。views Drug Discovery,2011,10(4):307-317.
[1]Cohen P.The origins of protein phosphorylation[J].Nat. Cell Biol.,2002,4(5):127-130.
[2]Manning G.Evolution of protein kinase signaling from yeast to man[J].Trends Biochem.Sci.,2002,27(10):514-520.
[3]Manning G.The protein kinase complement of the human genome[J].Science(Washington,DC,U.S).,2002,298:1 912-1 916,1 933-1 934.
[4]Davis M I.Comprehensive analysis of kinase inhibitor selectivity[J].Nature Biotechnology,2011,29(11):1 046-1 051.
[5]Uetrecht J P.New concepts in immunology relevant to idiosyncratic drug reactions:the“danger hypothesis”and innate immune system[J].Chemical Research in Toxicology,1999,12(5):387-395.
[6]Potashman M H,Duggan M E.Covalent modifiers:An orthogonal approach to drug[J]Design.J.Med.Chem.,2009,52(5):1 231-1 246.
[7]Singh J.The resurgence of covalent drugs[J].Nature Re-
[8]Honigberg L E Verner,Z Pan.Inhibitors of bruton's tyrosine kinase[J].Design.J.Med.Chem.,2010,59(3):1 231-1 246.
[9]Kumar S,R Agrawal.Next generation tyrosine kinase inhibitor(TKI):Afatinib.Recent patents on anti-cancer drug discovery[J].Nature Reviews Drug Discovery,2014,9(3):382-393.
[10]Hanke J H,Joseph P Gardner,Robert L Dow,et al. Discovery of a novel,potent,and src family-selective tyrosine kinase inhibitor Study of Lck-and FynT-dependent T cell activation[J].Journal of Biological Chemistry,1996,271(2):695-701.
[11]Lee Y,Shim H S,Rark M S,et al.High EGFR gene copy number and skin rash as predictive markers for EGFR tyrosine kinase inhibitors in patients with advanced squamous cell lung carcinoma[J].Clinical Cancer Research,2012,18(6):1 760-1 768.
[12]Clark J D,M E Flanagan,J B.Telliez.Discovery and development of janus kinase(JAK) inhibitors for inflammatory diseases:miniperspective[J].Journal of Medicinal Chemistry,2014,57(12):5 023-5 038.
[13]Luo J,B D Manning,L C Cantley.Targeting the PI3KAkt pathway in human cancer:rationale and promise[J]. Cancer cell,2003,4(4):257-262.
[14]Yuan W,R M Krug.Influenza B virus NS1 protein inhibits conjugation of the interferon(IFN) induced ubiquitin like ISG15 protein[J].The EMBO Journal,2001,20(3):362-371.
[15]Sagara T.3,5-disubstituted alkynylebenzene compound and salt thereof.US 2014034035[P],2014.
[16]Martinez A.Preclinical efficacy on GSK 3 inhibitors:Towards a future generation of powerful drugs[J].Medicinal Research Reviews,2008,28(5):773-796.
[17]Perez D I,Valle P,Concepcion P,et al.Switching reversibility to irreversibility in glycogen synthase kinase 3 inhibitors:clues for specific design of new compounds[J]. Journal of Medicinal Chemistry,2011,54(12):4 042-4 056.
[18]Farooqui M,Wiestner Z A.Ibrutinib for the treatment of chronic lymphocytic leukemia[J].Journal of Medicinal Chemistry,2013,65(12):4 089-4 095.
[19]Haber D A,N S Gray,J Baselga.The evolving war on cancer[J].Cell,2011,145(1):19-24.
[20]Barluenga S.In vivo efficacy of natural product inspired irreversible kinase inhibitors[J].Chem Bio Chem,2010,11(12):1 692-1 699.
[21]Dungo R T,G M Keating.Afatinib:first global approval[J]. Drugs,2013,73(13):1 503-1 515.
[22]Carmi C.Clinical perspectives for irreversible tyrosine kinase inhibitors in cancer[J].Biochemical Pharmacology,2012,84(11):1 388-1 399.◇
Advances in Irreversible Protein Kinase Inhibitors
ZHENG Peigen,ZHENG Chenglong
(Qingdao University of Science and Technology,Qingdao,Shandong 266042,China)
Protein kinases are a highly conserved enzyme systerm by a variety of enzymes,which are primarily responsible for the transfer of ATP Y-phosphate groups to tyrosine,serine,threonine and other amino acids.These amino acids play a role in signal transduction in cells.To date,the majority of listed kinase inhibitors are non-covalent ATP competitive inhibitors. But recently,the development of an irreversible inhibitors is favored by researchers,because of they can combine with cysteine or other nucleophilic amino acids residues to form covalent bonds in the ATP binding pocket.This article is mainly about the research progress in irreversible protein kinase inhibitors,and discussed the efficient development strategies of new covalent inhibitors.
protein kinase;covalent inhibitor;irreversibility
Q813
A
10.16693/j.cnki.1671-9646(X).2016.01.015
2015-09-13
郑培根(1989— ),男,硕士,研究方向为药物合成。
