腺苷酸激酶催化循环后期Mg2+转移的分子动力学模拟
2016-11-08崔大超任卫同李文飞南京大学物理学院南京微结构国家实验室人工微结构科学与技术协同创新中心南京210093
崔大超 任卫同 李文飞 王 炜(南京大学物理学院,南京微结构国家实验室,人工微结构科学与技术协同创新中心,南京210093)
腺苷酸激酶催化循环后期Mg2+转移的分子动力学模拟
崔大超任卫同李文飞王炜*,
(南京大学物理学院,南京微结构国家实验室,人工微结构科学与技术协同创新中心,南京210093)
腺苷酸激酶是一个包含三个结构域(LID结构域、NMP结构域和CORE结构域)的蛋白质分子,其主要作用是催化化学反应Mg2++ATP+AMP⇌2ADP+Mg2+,进而将细胞内ATP分子的浓度维持在合适的范围内。在腺苷酸激酶催化上述化学反应的过程中,需要有Mg2+的参与。最近的实验发现Mg2+不仅参与上述反应的化学步骤,而且对化学反应发生后底物的释放过程至关重要。已有晶体结构数据显示,在催化循环过程的化学反应步骤完成后,一个Mg2+可同时和分别位于LID结构域及NMP结构域的两个ADP分子配位。然而,在底物的释放与分离过程中,Mg2+可能只与其中一个ADP分子结合。由于Mg2+与ADP分子的结合情况会在很大程度上影响作为催化循环限速步骤的底物释放过程,因此人们有必要研究清楚在底物释放前Mg2+与催化产物ADP分子的配位情况,即Mg2+更倾向于与LID结构域的ADP分子结合还是与NMP结构域的ADP分子结合。本文中,我们对催化反应后底物释放前的酶-底物复合物(包含酶、两个ADP分子以及Mg2+)做了分子动力学模拟研究。我们基于metadynamics方法得到了Mg2+在两个ADP分子间转移的自由能面,发现在底物分离与释放过程中,Mg2+更倾向于与LID结构域的ADP分子结合。只有当LID结构域的ADP分子被质子化,同时NMP结构域的ADP分子处于去质子化状态时,Mg2+才会倾向于与NMP结构域的ADP分子结合。另外,我们也刻画了Mg2+转移过程中配体交换与脱水过程。本工作的研究结果有助于理解腺苷酸激酶催化循环后期的分子过程。
腺苷酸激酶;镁离子转移;Metadynamics;分子模拟
doi:10.3866/PKU.WHXB201511201
1 Introduction
It is well known that when biological systems perform their functions,various organic biomolecules,inorganic cofactors,such as Mg2+,Ca2+,and Zn2+,heme,and phosphate are involved and play vital roles1-4.The most typical examples are such that metal ions can mediate a number of biological processes,including catalysis, electron transport,and structure modulations and so on.To fully understand the related functioning mechanisms of biomolecules, it is important to study the molecular processes involved by such metal ions5-7.Adenylate kinase is a kind of important enzymes which can catalyze the reversible reaction Mg2++ATP+AMP⇌2ADP+Mg2+8.This enzyme contains three domains,namely the LID domain,NMP domain,and CORE domain(Fig.1(a)),and has been frequently used as a model system to study the protein conformational motions,protein folding,and the coupling between chemical reaction and protein conformational dynamics, both experimentally and computationally8-23.It is well known that the Mg2+is involved in the chemical step of the catalytic cycle. Recent experimental studies demonstrated that the Mg2+also plays a role of pivot during the phosphate transfer from ATP to AMP9. Particularly,the experimental data showed that the presence of Mg2+in the ADP binding site is crucial for the substrate releasing and the protein conformational opening9,which is the rate limit step of the catalytic cycle of the adenylate kinase24.Therefore, revealing the final Mg2+binding state of the substrate(i.e.,the two ADP molecules)before the substrate releasing is important for understanding the key factors which control the catalytic efficiency of the adenylate kinase.
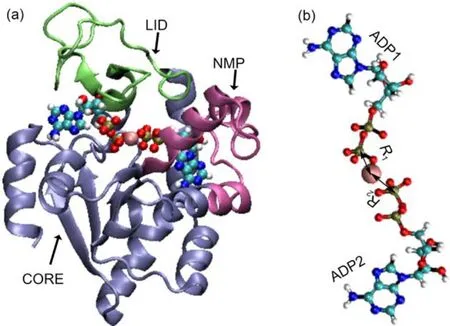
Fig.1 (a)Crystal structure of theAKE in the substrate bound conformation(pdb code:1AKE);(b)the initial conformation of the twoADPmolecules and the Mg2+
It was shown experimentally that the Mg2+coordinates to both ADP molecules right after the chemical step of the catalytic reaction(Fig.1(b))9.However,it is unclear whichADP molecule can bind with the Mg2+during the substrate releasing.As discussed in recent experimental work9,the Mg2+binding to the active site can promote the substrate releasing by modifying the local electrostatic interactions.Since the Mg2+binding to different ADP molecules can lead to very different perturbations to the local electrostatic environment,the information on the detailed Mg2+binding modes after the chemical step of the catalytic cycle is critical for understanding the factors controlling the substrate releasing and protein conformational opening.Due to the transient feature of the bound state of the post-catalytic complex,it is difficult to experimentally detect such information.Molecular dynamics simulations have been shown successful in revealing the molecular details of various complicated biological processes25-27.However, the conventional atomistic molecular dynamics is limited to simulatingthemolecularprocesseswithtimescaleslessthanmicrosecond,whichisusuallymuchshorterthantypicaltimescalesof functionalmotionsoflargeproteins.Forsuchcases,manybiased moleculardynamicsmethodsandenhancedsamplingmethodsare useful28-34.Metadynamicsisoneofthebiasedmolecular dynamics simulation methods,and has been extensively used in the studies of protein folding,functional motions,and transportation28,35-38.
In this work,by using metadynamics simulations,we simulated the Mg2+transfer between the two ADP molecules.From the simulation data,we constructed the free energy landscapes which can be used to characterize the Mg2+transfer to the individualADP molecules.Our results showed that the Mg2+prefers to coordinate with the ADP molecule of the LID domain.We found that only when the ADP of the LID domain was protonated,and simultaneously the ADP of the NMP domain was deprotonated,the Mg2+tended to coordinate with the ADP of the NMP domain.We also characterized the ligand exchange and dehydration processes of the Mg2+transfer.
2 Materials and methods
Our simulations started from the crystal structure of the adenylate kinase in E-coli(AKE)at the closed state with PDB code 1AKE39.In this structure,bis(adenosine)-5'-pentaphosphate(AP5) molecule was used to mimic the substrate.The initial structures of the two ADP molecules,which are the catalysis products,wereprepared by replacing the central phosphate group with Mg2+.The ADP molecules attached to the LID domain and NMP domain were denoted asADP1 andADP2,respectively.
In this work,the metadynamics simulations were used to accelerate the conformational sampling and to construct the free energy landscapes.In the metadynamics simulations,repulsive Gaussian potential was periodically added to the conformational space previously visited during a short time interval28,38.To characterize the conformational space,some collective variable(s) which are relevant to the interesting molecular processes need to be predefined.The deposited Gaussian potential tends to force the molecule escape from the energy basins,and therefore speeds up the sampling along the biologically relevant collective variable(s). Once all the basins are filled up by the Gaussian potential,the conformational motions of the proteins can cover the whole range of the relevant collective variable(s),and the accumulation of the filled Gaussian potential can be used to construct the free energy landscape.Previously,the metadynamics has been widely used in simulating the rare events of ion transportation,protein folding, and other biologically important processes28,35-38.
In order to monitor the Mg2+transfer between the two ADP molecules,we used the R1and R2,which correspond to the distances between the Mg2+and the Pβatoms of theADP1 andADP2, respectively,as the collective variables of the metadynamics simulations.The amplitude and width of the deposited Gaussian packages were set as 0.4187 kJ∙mol-1and 0.01 nm,respectively. The Gaussian packages were added with an interval of 2 ps.To get better sampling convergence,we introduced harmonic restraints to the lower and upper boundaries of the R1and R2.The lower and upper boundaries were set as 0.3 and 0.6 nm,respectively.The lower(upper)boundary corresponds to the distance at which the oxygen atom coordinates to the Mg2+(the oxygen atom is well separated from the Mg2+).Larger values of upper boundary are better,but can significantly extend the available conformational space,therefore increase the computational time.Due to the lower and upper boundaries,the obtained free energy landscapes can only cover the conformational spaces with R1and R2range from 0.3 to 0.6 nm,respectively,although smaller or larger values of R1and R2can be sampled due to the weak harmonic restraints.Here, the lower and upper boundaries were 0.3 nm.
The simulations were performed with NAMD2.9 software package40.The particle mesh Ewald algorithm was used with periodic boundary condition41.The direct term of the electrostatic interactions and the non-bonded van der Waals interactions were truncated at 1 nm.Firstly,the system was minimized by steepest descent method and conjugate gradient methods.Then the systems were gradually heated to 300 K with NVT ensemble and further relaxed at 300 K and 1.013×105Pa with NPT ensemble for 1.0 ns.The product metadynamics simulations were conducted for 100 ns.Due to the added biasing potential,with such short-length metadynamics simulations,we can observe molecular events of longer than microsecond time scale.
Since the ADP molecules can be protonated,it will be interesting to investigate how the protonation states of the two ADP molecules can affect the final Mg2+binding state.In this work, each ADP molecule can be either protonated or deprotonated. Since there are two ADP molecules in the simulation system,we totally conducted four simulations,including:(1)both ADP molecules were deprotonated(denoted by“DD”),(2)only ADP molecule of the LID domain was protonated(denoted by“D(H) D”),(3)only ADP molecule of the NMP domain was protonated (denoted by“DD(H)”),and(4)both ADP molecules were protonated(denoted by“D(H)D(H)”).The systems were solvated in a cubic TIP3P water box with the box size of~343 nm3.Then,Na+and Cl-were added to neutralize the simulation box and to mimic the salt concentration of 0.1 mol∙L-1.The CHARMM36 force field was used for the protein and theADP molecules42.The partial charges of the protonated ADP molecules were obtained from a webserver Paramchem43.The protein structure was visualized by VMD software44.
3 Results and discussion
Fig.2(a)shows the time series of the two collective variables, R1and R2(i.e.,the distances between the Mg2+and the Pβatoms of the ADP1 and ADP2,respectively).One can see that in the later stage of the metadynamics sampling,both collective variables can sample along wide range of the conformational space.The R1and R2hop between small values and large values,suggesting high sampling quality of the metadynamics simulations.With such metadynamics simulation data,we can construct the free energy landscape according to the deposited biasing potentials.The constructed free energy landscapes projected on the two collective variables are shown in Fig.2(b)at different stages of the metadynamics simulations for the conditions with two ADP molecules deprotonated.From the free energy landscape,we can see four major basins at most,which correspond to four Mg2+binding states,namely,Mg2+simultaneously binds to both ADP molecules (left-bottom),Mg2+binds toADP1 molecule(left-top),Mg2+binds toADP2 molecules(right-bottom),and Mg2+does not bind to any ADP molecules(right-top).We can see that the constructed free energy landscape does not change significantly after 60 ns except that a new conformational state at the right-top corner of the free energy landscape can appear with the simulation time.In this work,we mainly discuss the relative difference of the free energy values between the conformational states locating at the left-top corner and right-bottom corner of the free energy landscape,which gets converged after the simulation time of 60 ns.The representative snapshots of the major conformational states are also shown in Fig.2(c).Compared to the ADP2 bound state,the state with Mg2+binding to the ADP1 molecule has lower free energy,suggesting that after the catalysis reaction,the Mg2+prefers to bind with the ADP1 molecule before the substrate releasing when both ADP molecules are deprotonated.Such results are important because previous experimental data suggested that the Mg2+ions are involved in the substrate releasing and AKE domain opening by disrupting the local water environment and electrostatic in-teractions.Our results show that for the deprotonated ADP molecules,the Mg2+prefers to bind with theADP1 molecule,therefore will have more pronounced effect on the releasing of the ADP1 molecule.
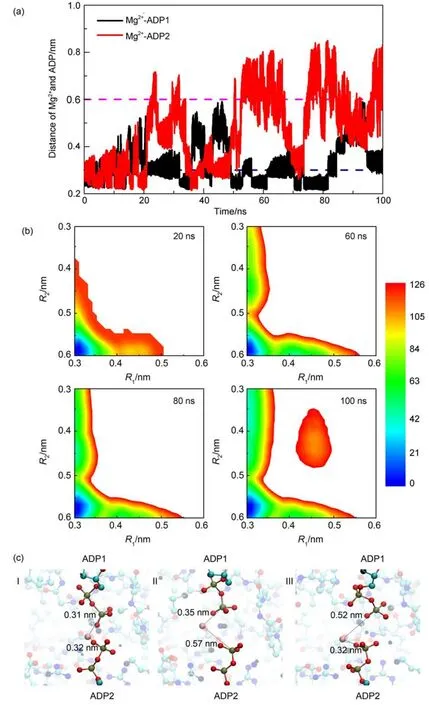
Fig.2 (a)Time series of the distances between the Mg2+and the Pβatoms of theADP1 andADP2,i.e.,the collective variables,R1and R2, respectively;(b)free energy landscapes projected on the R1and R2at different stages of the metadynamics simulations (the unit of the free energy is kJ∙mol-1);(c)represent snapshots showing the binding modes between the Mg2+and the twoADPmolecules in the three major states
In Fig.2(a),we can observe a state with the values of R1and/or R2around 0.25 nm,which is smaller than the lower boundary ofthe metadynamics simulations.As discussed in the previous section,sampling beyond the boundary is arising from the weak restraints of the boundaries in the metadynamics simulations.Due to the restraint in the metadynamics simulations,although the R1and R2can take the values smaller than the lower boundary,the biasing potentials were added to the conformational space within the boundaries.Consequently,the basins with R1and/or R2values of 0.3 nm in the free energy landscapes in Fig.2(b)actually also contain the states with the values of R1and/or R2lower than the lower boundary.However,such treatment does not affect our discussion since both the states with R1and/or R2values of 0.3 nm and the states with R1and/or R2values lower than the lower boundary are considered as Mg2+bound states in this work.
Next,we investigate how the protonation state affects the Mg2+transfer.We repeated the above metadynamics simulations for another three protonation states,namely,D(H)D,DD(H),and D(H)D(H).DD is both ADP molecules deprotonated,D(H)D is onlyADP1 protonated,DD(H)is onlyADP2 protonated,D(H)D (H)is both ADP1 and ADP2 protonated.The constructed free energy landscapes,together with the one without protonation are shown in Fig.3.For the protonation states DD(H)and D(H)D(H), the results are very similar to that of without protonation(Fig.2 (b)),namely,the Mg2+prefers to bind with the ADP1 after the catalysis.Only for the protonation state D(H)D,in which the ADP1 is protonated,and simultaneously,theADP2 is deprotonated,the Mg2+prefers to bind with the ADP2.Possible reason for such protonation effects on the Mg2+bound state is that the protonation introduces a unit of positive charge,which tends to have repulsive interactions between theADP1 and the Mg2+.As a result, the Mg2+has more probability to bind with theADP2 thanADP1.
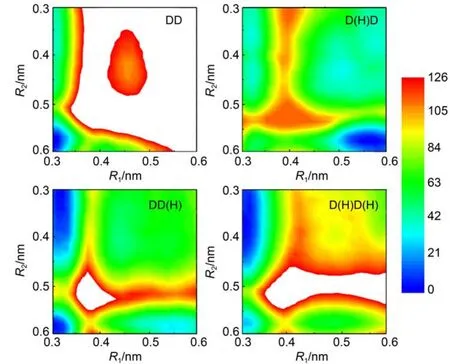
Fig.3 Free energy landscapes projected on the R1and R2for different protonation states
The Mg2+tends to coordinate with water molecules in solvent. In the crystal structure of AKE,the Mg2+can coordinate with protein residues,ADP molecules,and water molecules.From the catalytic transition state structure to the post-catalytic structure right before the substrate releasing,the coordination environment of Mg2+was modified.Particularly,the water molecules may exchange with theADP molecules.Fig.4(a)shows a representative trajectory,in which a Pβatom leaves the coordination shell of the Mg2+.At the same time,one water molecule comes into the coordination shell.Such result suggests the ligand exchange process during the Mg2+transfer.Since the breaking up of coordination bond needs to overcome a high energy barrier,the simultaneous coordination of a water molecule,namely ligand exchange,may help to reduce the energy barrier,therefore speed up the Mg2+transfer.Such hydration and/or dehydration of metal ions during the folding of other metalloprotein have also been observed in molecular dynamics simulations,and was shown to play crucial roles in speeding up the protein conformational dynamics2.
Recent experimental data suggested that the Mg2+binding to the ADP molecules may affect the local water structure and dynamics, therefore contribute to the substrate releasing9.In this work,we also calculated the averaged number of water molecules coordinated to the Mg2+at different conformational states characterized by R1and R2.If we assume that the number of water molecules coordinated to the Mg2+is determined by the values of R1and R2, namely,the R1and R2are good reaction coordinates to define the local structure of Mg2+binding site,we can calculate the averaged number of water molecules bound to the Mg2+directly from the metadynamics simulations.Fig.4(b)shows the averaged number of water molecules coordinated to the Mg2+at different conformational states defined by the R1and R2.One can see that for all the four protonation states,different Mg2+bound states have different hydration extents.For the state with two ADP molecules bound to the Mg2+,the number of coordinated water molecules is the lowest,which is reasonable because the maximum of the Mg2+coordination number is fixed.Consequently,coordination of twoADP molecules will reduce the number of coordinated water molecules.With the breaking up of one of the ADP-Mg2+coordination bonds,around one or two water molecules may enter the coordination shell.When both ADP-Mg2+coordination bonds are broken,up to five water molecules can enter the coordination shell.These results suggested that the change of the Mg2+bound state can change the hydration state of the active site,therefore may contribute to the modulation of the substrate releasing steps.
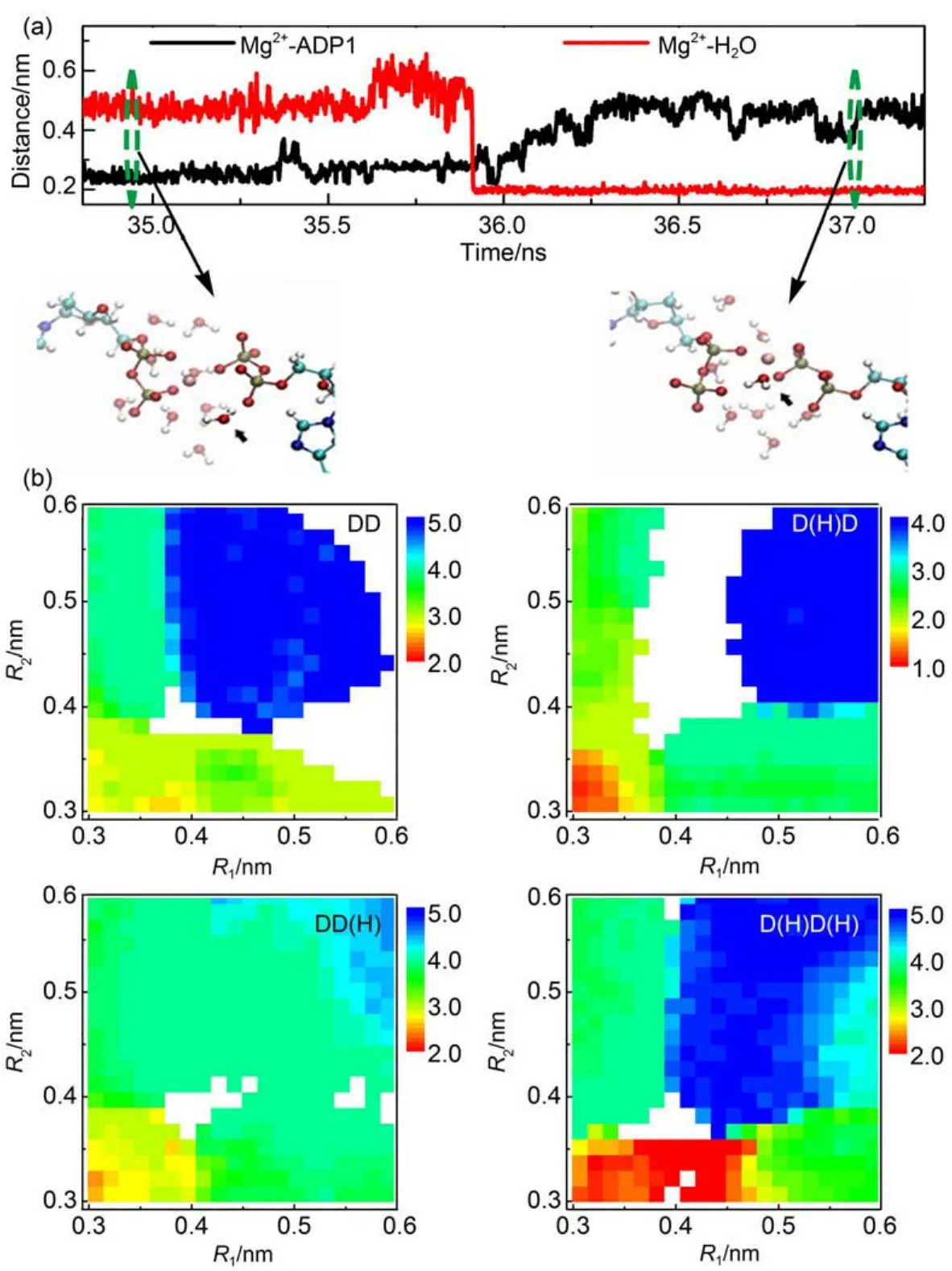
Fig.4 (a)Representative trajectory for the distances between Mg2+and Pβatom ofADP1 and between Mg2+and O atom of water molecule as a function of time;(b)averaged number of water molecules coordinated with Mg2+at different conformational states represented by collective variables R1and R2for the four protonation states
In Fig.4(a),the representative snapshots at two time points are also shown.color on web version.
4 Conclusions
Insummary,byusingmetadynamicssimulations,westudiedthe Mg2+transfer around the substrate binding site of the adenylate kinaseinthelatestageofthecatalyticcycle.Ourresultsshowthat theMg2+tendstobindwiththeADP1afterthechemicalstepofthe catalysisinmostoftheprotonationstates.Onlyfortheprotonation statewithADP1protonated,andsimultaneouslyADP2deprotonated,theMg2+haspreferencetobindwiththeADP2moleculesdue to the electrostatic repulsion introduced by the protonation. Meanwhile,wedemonstratedtheligandexchangebetweenwater moleculesandPβatomofADPduringtheMg2+transfer.Inaddition, theresultsshowthattheMg2+transferchangesthehydrationstateof theMg2+,whichmayinturnaffectthewaterstructureanddynamics around the substrate binding pocket of theAKE.Our simulation resultsareimportantforunderstandingthefunctionalmechanism ofkinaseinvolvingMg2+ions.Firstly,werevealthepossibleeffects ofADPprotonationontheMg2+transferduringthelatestageofthe AKEcatalyticcycle.Secondly,weshowthedetailedMg2+binding modes after the chemical step of the catalytic cycle,which is importanttounderstandtheelectrostaticroleofMg2+forthesubstrate releasing.Thirdly,weprovidemoredetailedstructuralanddynamics information of the active site of theAKE,which is important for understandingitsfunctioningmechanism.ThemolecularmechanismsofsubstratereleasingconsideringdifferentMg2+boundstates deservefurtherworks.
References
(1)Palm-Espling,M.E.;Niemiec,M.S.;Wittung-Stafshede,P. Biochim.Biophys.Acta 2012,1823,1594.doi:10.1016/j. bbamcr.2012.01.013
(2)Li,W.;Zhang,J.;Wang,J.;Wang,W.J.Am.Chem.Soc.2008, 130,892.doi:10.1021/ja075302g
(3)Alberts,B.;Johnson,A.;Lewis,J.;Raff,M.;Roberts,K.; Walter,P.Molecular Biology of the Cell,1st ed.;Garland Science,Taylor&Francis Group:New York,2007.
(4)Wilson,C.J.;Apiyo,D.;Wittung-Stafshede,P.Q.Rev. Biophys.2004,37,285.
(5)Li,W.;Wang,W.;Takada,S.Proc.Natl.Acad.Sci.U.S.A. 2014,111,10550.doi:10.1073/pnas.1402768111
(6)Li,W.;Zhang,J.;Su,Y.;Wang,J.;Qin,M.;Wang,W.J.Phys. Chem.B 2007,111,13814.doi:10.1021/jp076213t
(7)Li,W.;Wang,J.;Zhang,J.;Wang,W.Curr.Opin.Struct.Biol. 2015,30,25.doi:10.1016/j.sbi.2014.11.006
(8)Muller,C.W.;Schlauderer,G.J.;Reinstein,J.;Schulz,G.E. Structure 1996,4,147.doi:10.1016/S0969-2126(96)00018-4
(9)Kerns,S.J.;Agafonov,R.V.;Cho,Y.J.;Pontiggia,F.;Otten, R.;Pachov,D.V.;Kutter,S.;Phung,L.A.;Murphy,P.N.; Thai,V.;Alber,T.;Hagan,M.F.;Kern,D.Nat.Struct.Mol. Biol.2015,22,124.doi:10.1038/nsmb.2941
(10)Formoso,E.;Limongelli,V.;Parrinello,M.Sci.Rep.2015,5, 8425.doi:10.1038/srep08425
(11)Giri Rao,V.V.;Gosavi,S.PLoS Comp.Biol.2014,10, e1003938.
(12)Wang,Y.;Gan,L.F.;Wang,E.K.;Wang,J.J.Chem.Theory Comput.2013,9,84.doi:10.1021/ct300720s
(13)Li,W.;Terakawa,T.;Wang,W.;Takada,S.Proc.Natl.Acad. Sci.U.S.A.2012,109,17789.doi:10.1073/pnas.1201807109
(14)Pirchi,M.;Ziv,G.;Riven,I.;Cohen,S.S.;Zohar,N.;Barak, Y.;Haran,G.Nat.Commun.2011,2,493.doi:10.1038/ ncomms1504
(15)Li,W.;Wolynes,P.G.;Takada,S.Proc.Natl.Acad.Sci.U.S. A.2011,108,3504.doi:10.1073/pnas.1018983108
(16)Daily,M.D.;Phillips,G.N.,Jr.;Cui,Q.J.Mol.Biol.2010, 400,618.doi:10.1016/j.jmb.2010.05.015
(17)Schrank,T.P.;Bolen,D.W.;Hilser,V.J.Proc.Natl.Acad.Sci. U.S.A.2009,106,16984.doi:10.1073/pnas.0906510106
(18)Beckstein,O.;Denning,E.J.;Perilla,J.R.;Woolf,T.B. J.Mol.Biol.2009,394,160.doi:10.1016/j.jmb.2009.09.009
(19)Lu,Q.;Wang,J.J.Am.Chem.Soc.2008,130,4772.doi: 10.1021/ja0780481
(20)Whitford,P.C.;Miyashita,O.;Levy,Y.;Onuchic,J.N.J.Mol. Biol.2007,366,1661.doi:10.1016/j.jmb.2006.11.085
(21)Henzler-Wildman,K.A.;Lei,M.;Thai,V.;Kerns,S.J.; Karplus,M.;Kern,D.Nature 2007,450,913.doi:10.1038/ nature06407
(22)Bae,E.;Phillips,G.N.,Jr.Proc.Natl.Acad.Sci.U.S.A.2006, 103,2132.doi:10.1073/pnas.0507527103
(23)Miyashita,O.;Onuchic,J.N.;Wolynes,P.G.Proc.Natl.Acad. Sci.U.S.A.2003,100,12570.doi:10.1073/pnas.2135471100
(24)Wolf-Watz,M.;Thai,V.;Henzler-Wildman,K.;Hadjipavlou, G.;Eisenmesser,E.Z.;Kern,D.Nat.Struct.Mol.Biol.2004, 11,945.doi:10.1038/nsmb821
(25)Ma,W.;Schulten,K.J.Am.Chem.Soc.2015,137,3031.doi: 10.1021/ja512605w
(26)Liu,F.F.;Dong,X.Y.;Sun,Y.Acta Phys.-Chim.Sin.2010, 26,1643.[刘夫锋,董晓燕,孙彦.物理化学学报,2010,26, 1643.]doi:10.3866/PKU.WHXB20100613
(27)Lindorff-Larsen,K.;Piana,S.;Dror,R.O.;Shaw,D.E. Science 2011,334,517.doi:10.1126/science.1208351
(28)Laio,A.;Gervasio,F.L.Rep.Prog.Phys.2008,71,126601.
(29)Darve,E.;Rodriguez-Gomez,D.;Pohorille,A.J.Chem.Phys 2008,128,144120.doi:10.1063/1.2829861
(30)Warmflash,A.;Bhimalapuram,P.;Dinner,A.R.J.Chem. Phys.2007,127,154112.doi:10.1063/1.2784118
(31)Terakawa,T.;Takada,S.Biophys.J.2011,101,1450.doi: 10.1016/j.bpj.2011.08.003
(32)Li,W.;Yoshii,H.;Hori,N.;Kameda,T.;Takada,S.Methods 2010,52,106.doi:10.1016/j.ymeth.2010.04.014
(33)Li,W.;Takada,S.J.Chem.Phys.2009,130,214108.doi: 10.1063/1.3146922
(34)Li,W.;Takada,S.Biophys.J.2010,99,3029.doi:10.1016/j. bpj.2010.08.041
(35)Bussi,G.;Gervasio,F.L.;Laio,A.;Parrinello,M.J.Am. Chem.Soc.2006,128,13435.doi:10.1021/ja062463w
(36)Laio,A.;Parrinello,M.Proc.Natl.Acad.Sci.U.S.A.2002, 99,12562.doi:10.1073/pnas.202427399
(37)Pfaendtner,J.;Branduardi,D.;Parrinello,M.;Pollard,T.D.; Voth,G.A.Proc.Natl.Acad.Sci.U.S.A.2009,106,12723. doi:10.1073/pnas.0902092106
(38)Gervasio,F.L.;Parrinello,M.;Ceccarelli,M.;Klein,M.L. J.Mol.Biol.2006,361,390.doi:10.1016/j.jmb.2006.06.034
(39)Muller,C.W.;Schulz,G.E.J.Mol.Biol.1992,224,159.doi: 10.1016/0022-2836(92)90582-5
(40)Phillips,J.C.;Braun,R.;Wang,W.;Gumbart,J.;Tajkhorshid, E.;Villa,E.;Chipot,C.;Skeel,R.D.;Kale,L.;Schulten,K. J.Comput.Chem.2005,26,1781.
(41)Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98, 10089.doi:10.1063/1.464397
(42)Huang,J.;MacKerell,A.D.,Jr.J.Comput.Chem.2013,34, 2135.doi:10.1002/jcc.23354
(43)Vanommeslaeghe,K.;Raman,E.P.;MacKerell,A.D.,Jr. J.Chem.Inf.Model.2012,52,3155.doi:10.1021/ci3003649
(44)Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graphics 1996, 14,33.doi:10.1016/0263-7855(96)00018-5
Metadynamics Simulations of Mg2+Transfer in the Late Stage of the Adenylate Kinase Catalytic Cycle
CUI Da-ChaoREN Wei-TongLI Wen-Fei§WANG Wei*,§
(National Laboratory of Solid State Microstructure,Department of Physics,Collaborative Innovation Center of Advanced Microstructures,Nanjing University,Nanjing 210093,P.R.China)
Adenylate kinase is a kind of important enzymes which can catalyze the reversible reaction Mg2++ATP+AMP⇌2ADP+Mg2+where the Mg2+coordination around the active site plays a crucial role.It was shown experimentally that one Mg2+ion can coordinate to both ADP molecules right after the chemical step of the catalytic reaction.During the substrate releasing and separation,the Mg2+may transfer to one of the ADP molecules.However,it is unclear which ADP molecule binds with the Mg2+during the substrate releasing.In this work,by using metadynamics method,we conducted molecular simulations on the adenylate kinase complexed with two ADP molecules and one Mg2+,which corresponds to the postcatalysis enzyme-substrate complex.We constructed the free energy landscapes characterizing the Mg2+transfer to the individual ADP molecules.Our results show that the Mg2+has preference to attach with the ADP molecule of the LID domain.We found that only when the LID domain ADP is protonated,andsimultaneously the NMP domain ADP is deprotonated,the Mg2+tends to attach with the NMP domain ADP. We also characterized the ligand exchange and dehydration processes during the Mg2+transfer.Our results provide insights into the molecular process during the late state of the adenylate kinase catalytic cycle.
Adenylate kinase;Mg2+transfer;Metadynamics;Molecular simulation
July 28,2015;Revised:November 13,2015;Published on Web:November 20,2015.
O641
*Corresponding author.Email:wfli@nju.edu.cn,wangwei@nju.edu.cn.
§These authors contributed equally to this work.
The project was supported by the National Natural Science Foundation of China(11334004,81421091).
国家自然科学基金(11334004,81421091)资助项目
©Editorial office ofActa Physico-Chimica Sinica
