替硝唑有关物质检查方法改进
2016-11-07尼珍边巴卓玛阿萍
尼珍,边巴卓玛,阿萍
(西藏自治区食品药品检验所,西藏拉萨850000)
替硝唑有关物质检查方法改进
尼珍,边巴卓玛,阿萍
(西藏自治区食品药品检验所,西藏拉萨850000)
目的改进替硝唑有关物质的检查方法。方法使用Zorbax SB-C8柱(250 mm×3.0 mm,5 m),流动相为水-甲醇-乙腈(83∶7∶10),检测波长为320 nm,流速为0.5 mL/min,柱温为35℃,进样量为20 L。结果在此色谱条件下,替硝唑与有关物质均能完全分离;替硝唑有关物质A、甲硝唑及替硝唑有关物质B的质量浓度在1~100 g/mL(r=1.000 0)范围内与峰面积线性关系良好;替硝唑杂质A、甲硝唑及替硝唑杂质B的检测限分别为0.04,0.20,0.20 ng;杂质A的平均回收率为96.23%,RSD为1.52%(n=6);甲硝唑的平均回收率为95.34%,RSD为1.64%(n=6);杂质B的平均回收率为96.72%,RSD为1.33%(n=6)。结论该方法简便,准确,专属性强,可用于替硝唑有关物质检查。
高效液相色谱法;替硝唑;有关物质
替硝唑是一种抗厌氧菌及抗原虫感染的药物,是继甲硝唑后疗效更佳、疗程更短、耐受性更好、体内分布更广的硝咪唑类衍生物,于20世纪60年代后期由美国辉瑞公司研究开发,商品名为Fasigyn,1972年在瑞士和西德上市,1991年《日本药局方》12版改正版已收载(JP 557页)。1993年,湖北省医药工业研究所率先在国内独家研制成功,并取得了卫生部颁发的新药证书[93(卫药证字X-65)号]。同年,丽珠制药厂替硝唑片(丽珠快服净)获卫生部颁发的生产批准文号[93(卫药试字X-28-1)号][1-3]。2010年版《中国药典》(ChP 2010)、《日本药局方》16版(JP 16)、《欧洲药典》7.0版(EP 7.0)、《英国药典》2013版(BP 2013)及《美国药典》35版(USP 35)均收载了替硝唑,但对有关物质的检查方法均不相同。ChP 2010、EP 7.0和BP 2013中均采用高效液相色谱(HPLC)法,USP 35和JP 16采用薄层色谱(TLC)法。本研究中借鉴EP 7.0中该品种的杂质控制相关信息,建立了系统适用性更强、分离度更好的替硝唑有关物质检查方法,为进一步完善和提高《中国药典》替硝唑有关物质检查方法提供参考。
1 仪器与试药
1.1仪器
E2695型液相色谱仪,2998紫外检测器,Empower色谱工作站(Waters公司)。
1.2试药
替硝唑杂质A对照品(中国食品药品检定研究院,批号为100512-200802,含量为99.8%);甲硝唑对照品(中国食品药品检定研究院,批号为100191-200606,含量为99.9%);替硝唑杂质B对照品(浙江苏泊尔制药有限公司,批号为B001-001,含量为99.7%);甲醇、乙腈为色谱纯,水为纯化水;14批替硝唑原料分别由浙江苏泊尔制药有限公司和印度Aarti Rrugs Limited公司提供。
2 方法与结果
2.1色谱条件
色谱柱:Zorbax SB-C8柱(250 mm×3.0 mm,5 μm);流动相:水-甲醇-乙腈(83∶7∶10);检测波长:320nm;流速:0.5mL/min;柱温:35℃;进样量:20 μL。
2.2溶液制备
供试品溶液:取样品(批号为001-130963)约10.0 mg,精密称定,置10 mL容量瓶中,加甲醇2 mL溶解,加流动相至刻度,摇匀,即得。
对照品溶液a:取供试品溶液1.0 mL,置100 mL容量瓶中,加流动相至刻度,摇匀,即得。
对照品溶液b:称取替硝唑杂质A对照品、甲硝唑对照品和替硝唑杂质B对照品各约5.0 mg,精密称定,置100 mL容量瓶中,加甲醇10 mL溶解,加流动相至刻度,摇匀,取此溶液2.0 mL置10 mL容量瓶中,加流动相至刻度,摇匀,即得。本溶液作为系统适用性溶液[2]。
2.3方法学考察
专属性试验:精密吸取2.2项下对照品溶液b、供试品溶液和空白对照溶液(流动相)各20 μL,按拟订色谱条件分别进样测定,结果供试品溶液在与对照品溶液相同保留时间处有相应色谱峰,而空白对照溶液则无此色谱峰,表明无干扰,见图1。
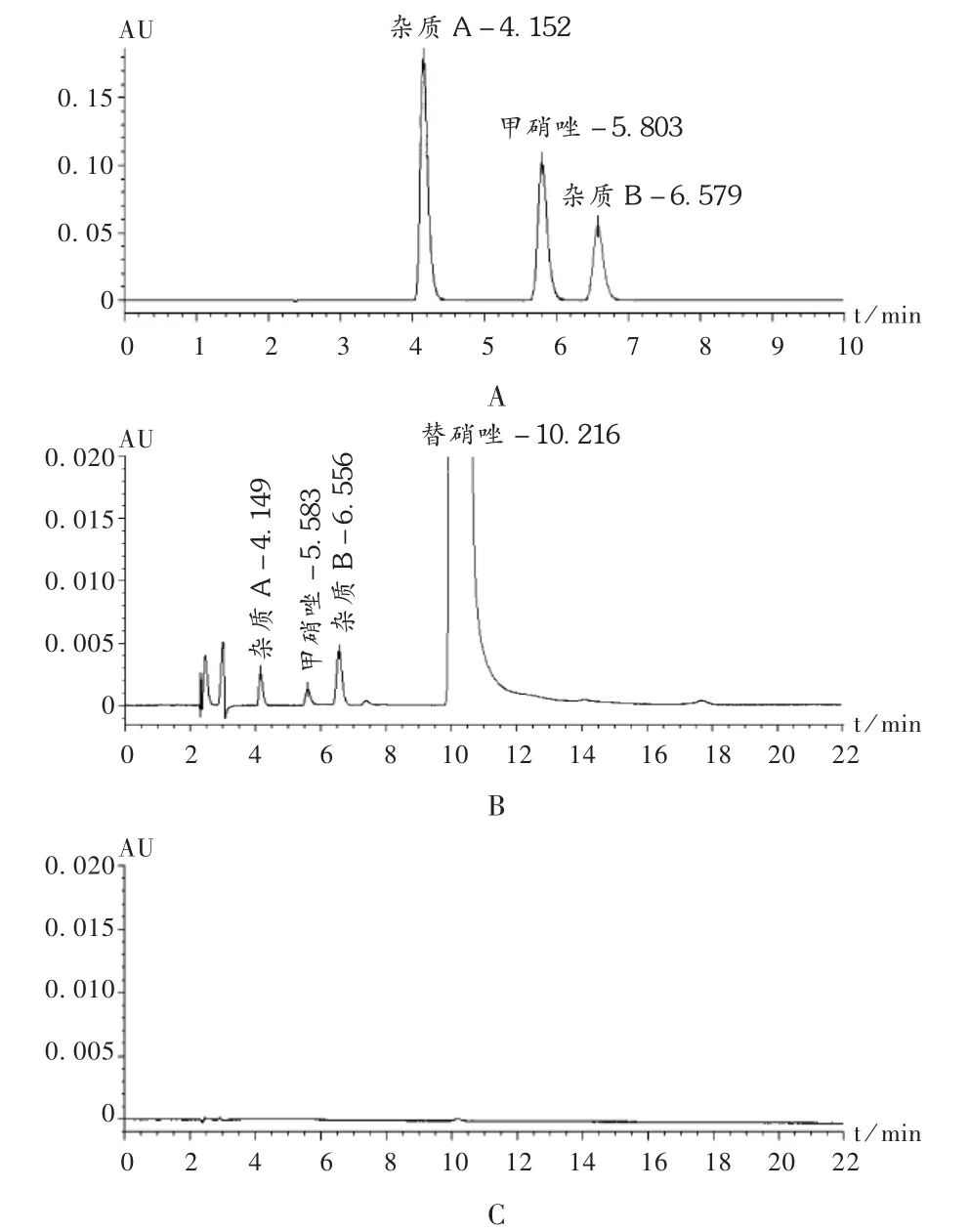
图1 高效液相色谱图
线性关系考察:精密称取替硝唑杂质A对照品、甲硝唑对照品和替硝唑杂质B对照品适量,用流动相配制系列质量浓度的混合对照品溶液1,5,10,20,40,80,100 μg/mL,按拟订色谱条件分别精密进样20 μL,记录色谱图。以各对照品溶液进样质量浓度(X,μg/mL)为横坐标,以峰面积(Y)为纵坐标,进行线性回归,得回归方程:替硝唑杂质A为Y=125 588 X+9 138.3,r= 1.000 0;甲硝唑为Y=99 542 X-865.2,r=1.000 0;替硝唑杂质B为Y=55 168 X-2 949.1,r=1.000 0。结果表明,各杂质质量浓度在1~100 μg/mL范围内与峰面积线性关系良好。
检测限测定:取“线性关系考察”项下质量浓度为10 μg/mL的对照品溶液,用稀释法测定检测限。按S/N=3计算,替硝唑杂质A、甲硝唑及替硝唑杂质B的检测限分别为0.04,0.20,0.20 ng。
精密度试验:精密吸取2.2项下对照品溶液b20 μL,连续进样6次。结果杂质A峰面积的RSD为0.14%(n=6),甲硝唑峰面积的RSD为0.16%(n=6),杂质B峰面积的RSD为0.13%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取2.2项下供试品溶液,分别于0,4,8,12,16,20,36,48 h进样20 μL,记录峰面积。结果杂质A峰面积的RSD为0.26%(n=8),甲硝唑峰面积的RSD为0.39%(n=8),杂质B峰面积的RSD为0.58%(n=8),表明供试品溶液在48 h内较稳定。
耐用性试验:精密吸取2.2项下供试品溶液,用2个不同品牌的色谱柱,即资生堂UG120 C8柱(250 mm× 4.6 mm,5 μm)和安捷伦Zorbax SB-C8柱(250 mm× 3.0 mm,5 μm),按拟订色谱条件分别精密进样20 μL。结果杂质A峰面积的RSD为1.46%,甲硝唑峰面积的RSD为1.47%,杂质B峰面积的RSD为0.97%,表明该方法系统耐用性较好。
加样回收试验:精密称取已知含量杂质A、甲硝唑及杂质B的样品(批号为001-130963,杂质A、甲硝唑及杂质B的含量分别为0.02%,0.01%,0.09%)约5.0 mg,共6份,分别精密加入含杂质A、甲硝唑及杂质B的混合对照品溶液(质量浓度杂质A为1.02 μg/mL,甲硝唑为0.51 μg/mL,杂质B为4.48 μg/mL)1.0 mL,按2.2项下方法制备供试品溶液,按拟订色谱条件分别精密进样20 μL,计算回收率,结果见表1。
2.4有关物质含量测定
按2.2项下方法制备各供试品溶液,按拟订色谱条件分别精密进样20 μL,按外标法(用于杂质A、甲硝唑及杂质B)和自身对照法(用于其他杂质)计算有关物质含量。结果见表2。
3 讨论
3.12种有关物质检查方法的比较
ChP 2010和EP 7.0均采用高效液相色谱法。ChP 2010中只检查非特定杂质,EP 7.0中检查特定杂质A、杂质B及其他非特定杂质。使用EP 7.0方法测定的供试品溶液色谱图中杂质峰的数量较多且分离度较好。另外,ChP 2010中供试品溶液和对照品溶液制备使用流动相溶解,而流动相含80%水系,替硝唑本身几乎不溶于水,造成溶解困难;EP 7.0中供试品溶液和对照品溶液制备使用甲醇溶解,替硝唑略溶于甲醇,故由溶解不完全造成的误差较小。ChP 2010中流动相使用0.05%磷酸二氢钾溶液-甲醇(80∶20),EP 7.0中使用乙腈-甲醇-水(10∶20∶70),此流动相对色谱柱损伤相对较小,能延长其使用寿命。
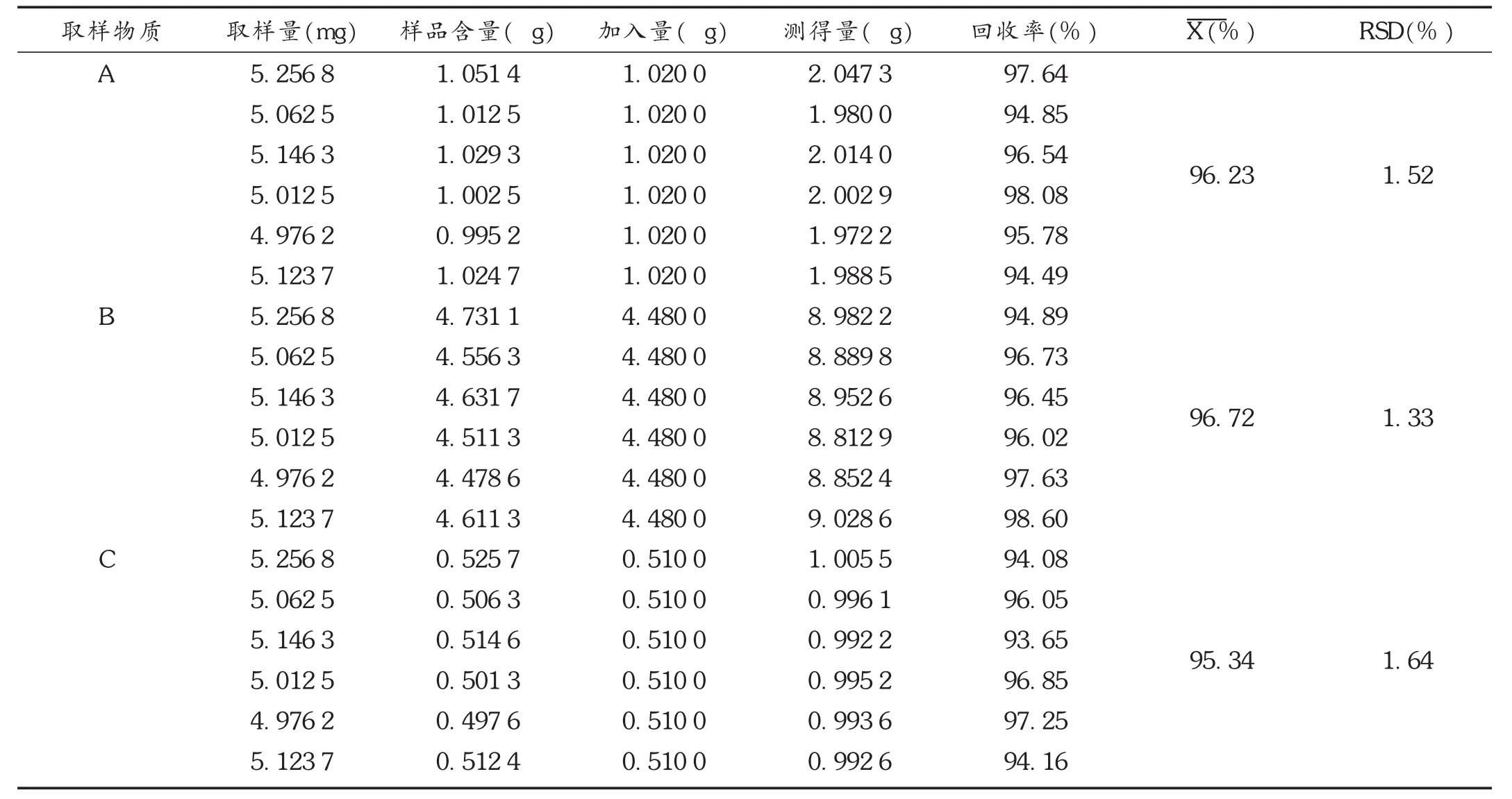
表1 加样回收试验结果(n=6)

表2 有关物质含量测定结果(%)
3.2供试品溶液质量浓度确定
EP 7.0中替硝唑供试品溶液的质量浓度为0.1 mg/mL,此质量浓度太低造成杂质峰面积积分不准确,故本研究中供试品溶液的质量浓度定为1.0 mg/mL。3.3系统适用性溶液制备
目前,大都采用2种基本原料合成替硝唑,即以2-甲基-5-硝咪唑或甲硝唑为原料合成替硝唑[4-6]。替硝唑杂质A和杂质B来源明确,分别为起始物料2-甲基-5-硝咪唑和反应副产物1-[2-(乙基磺酰基)乙基]-2-甲基-4-硝基-1H-咪唑。EP 7.0中以杂质A和杂质B的混合对照溶液作为系统适用性溶液,但未考虑甲硝唑,而在样品中除检出杂质A和杂质B外,也检出甲硝唑,我们认为可将甲硝唑列入替硝唑的特定杂质检查中。故制备了2.2项下含杂质A、甲硝唑和杂质B的混合对照溶液,并作为系统适用性溶液。
3.4流动相选择
当使用EP 7.0中流动相比例水-甲醇-乙腈(70∶20∶10)时,2.2项下的系统适用性溶液只检出2个峰,表明该流动相的分离度不好。调节流动相比例至水-甲醇-乙腈(80∶10∶10)后,各组分得以分离,但甲硝唑与杂质B分离度为1.8,不太理想。调节流动相比例至水-甲醇-乙腈(83∶7∶10)后,甲硝唑与杂质B分离度达到2.7,能够接受。
综上所述,改进后的方法能很好地用于替硝唑有关物质的检查,且方法中规定的各特定有关物质与替硝唑合成工艺相关,能较好地反映工艺过程及工艺稳定性。
[1]胡葆诚,曹筱秀.国内上市的5-硝基咪唑药物评价[J].医药导报,1995,14(2):75-76.
[2]杨阳.替硝唑[J].中国药理学通报,1994,10(5):386.
[3]杜达耿.替硝唑简介[J].中国医院药学杂志,1994,14(5):232.
[4]张珩,陈中,杨艺虹,等.替硝唑合成工艺的改进[J].精细化工,2002,19(6):332-333.
[5]郭春.替硝唑合成路线图解[J].中国医药工业杂志,1996,27(7):336.
[6]杨艺虹,杨建设,张珩,等.替硝唑合成路线评述[J].武汉化工学院学报,2001,23(1):31-33.
Improvement Method for Tinidazole Related Substances
Nizhen,Bianbazhuoma,Aping
(Tibet Institute for Food and Drug Control,Lhasa,Tibet,China850000)
ObjectiveTo improve the method for Tinidazole related substances.MethodsZorbax SB-C8(250 mm×3.0 mm,5 μm)column was used with a mobile phase of water-methanol-acetonitrile(83∶7∶10).The flow rat was 0.5 mL/min with column temperature at 35℃,the wavelength of detect was set at 320 nm.ResultsUnder the established chromatographic conditions,tinidazole and the related substances were separated completely.The standard curves obtained from impurity A,metronidazole and impurity B were linear within the range of 1-100 μg/mL with the correlation coefficient of r=1.000 0,the limits of detection were 0.04,0.20,0.20 ng,the average recovery rate of impurity A was 96.23%,RSD=1.52%(n=6),the average recovery rate of metronidazole was 95.34%,RSD=1.64%(n=6),and the average recovery rate of impurity B was 96.72%,RSD=1.33%(n=6).ConclusionThe established method is simple,accurate,specific and can be used for determination related substances of tinidazole.
HPLC;tinidazole;related substances
R927.11;R978.61
A
1006-4931(2016)18-0070-04
尼珍(1975-),女,副主任药师,主要从事食品药品检验工作,(电话)0891-6813172(电子信箱)nizhen1221@163.com。
(2016-03-18;
2016-05-26)
