钠盐溶液中U(Ⅵ)的电化学电子转移与晶化研究
2016-11-03宗美荣何辉超董发勤孙仕勇刘明学聂小琴
宗美荣, 何辉超, 董发勤,, 何 平, 孙仕勇, 刘明学, 聂小琴
(1. 西南科技大学固体废物处理与资源化教育部重点实验室, 绵阳 621010;2. 西南科技大学四川省非金属复合与功能材料重点实验室-省部共建国家重点实验室培育基地, 绵阳 621010)
钠盐溶液中U(Ⅵ)的电化学电子转移与晶化研究
宗美荣1, 何辉超2, 董发勤1,2, 何平2, 孙仕勇1, 刘明学1, 聂小琴1
(1. 西南科技大学固体废物处理与资源化教育部重点实验室, 绵阳 621010;2. 西南科技大学四川省非金属复合与功能材料重点实验室-省部共建国家重点实验室培育基地, 绵阳 621010)
采用循环伏安法分析钠盐溶液中U(Ⅵ)的电化学行为, 恒电位电化学还原处理U(Ⅵ), 利用交流阻抗谱分析电化学还原反应中的过程动力学特性, 利用X射线衍射、 扫描电子显微镜和电子能谱等方法分析了U(Ⅵ)的电化学晶化. 结果表明, 在钠盐溶液中, U(Ⅵ)可通过电化学反应先还原成低价的U(V)并进一步还原为U(Ⅳ), U(Ⅳ)一步氧化为U(Ⅵ), U(Ⅳ)/U(Ⅵ)之间的电化学转化过程受扩散控制, 且U(Ⅵ)的电化学电子转移易受环境pH值的影响; 恒电位还原4 h时, 溶液中U(Ⅵ)的去除率可达90%, U(Ⅵ)的结晶固化产物主要以固态的(UO2)6O2(OH)8·6H2O(水铀矿) 和UO2的形式附着在工作电极上.
铀(Ⅵ); 电化学; 电化学阻抗谱; 结晶
四价铀在水溶液中易与其它离子基团形成络合物, 六价铀(U(Ⅵ))可溶性较好, 不易去除, 对人体健康和生态环境具有重大威胁[1,2]. 根据含铀废水的特点, 其处理方法及工艺主要有生物或化学吸附法、 沉淀法、 矿石还原法、 萃取法、 蒸发浓缩法、 离子交换法、 零价铁处理法等[3~13], 但这些处理方法各有应用特点及范围, 存在一定的局限性.
电化学法可以有效表征化学反应过程中的电子转移[14]. 研究发现, U(Ⅵ)在一定电势下可被还原转化为低价态铀[15]:
(1)
(2)
目前, 已有U(Ⅵ)在不同种类离子液体中电子转移规律特性的报道. 如Anderson等[16]提出U(Ⅵ)在酸性AlCl3-EMIC(EMIC=氯化1-甲基-3-乙基咪唑)离子液体中的电化学还原过程为U(Ⅵ)→U(V)→U(Ⅳ)→U(III); Ikeda等[17]在[EMI]2[UO2Cl4]-咪唑氯盐离子溶液(EMI=1-乙基-3-甲基咪唑)中发现U(Ⅵ)与U(V)之间存在可逆氧化还原过程; 张秋月等[18]和王悦等[19]发现在丁基甲基咪唑氯盐和双三氟甲基磺酰亚胺三甲基丁基季铵盐离子液体中, U(Ⅵ)主要经历两步单电子还原过程转化为U(Ⅳ), 而U(Ⅳ)一步氧化为U(Ⅵ); 谭绪凤等[20]研究了U(Ⅵ)在季铵盐三甲基丙基铵双三氟甲基磺酰亚胺盐(N1113NTf2)离子液体中的电化学行为, 指出大量的U(Ⅵ)在电极表面可直接还原成为U(Ⅳ), 并进一步转化生成U(III), U(III)不稳定, 易与环境中的氧结合, 最终的产物为UO2. 然而, 对于水溶液钠盐体系中U(Ⅵ)的电子转移并无相关研究. 现含铀废水主要为核工业废水、 铀矿山废水和海水, 此3种体系均为简单的水溶液体系.
本文研究分析了钠盐体系中U(Ⅵ)向低价态转化的电化学电子转移规律和U(Ⅵ)在不同pH值NaCl水溶液中的存在形态, 并根据不同价态铀的电化学转化规律, 研究了电化学还原处理含U(Ⅵ)水溶液的工艺条件、 过程动力学特点及具体效果.
1 实验部分
1.1试剂
硝酸铀酰[UO2(NO3)2·6H2O, Ⅱ级分析纯, 北京化工厂]; 氯化钠和氢氧化钠(分析纯, 天津市大茂化学试剂厂); 盐酸(分析纯, 12 mol/L, 成都市联合化工试剂研究所); 偶氮胂Ⅲ和硝酸(分析纯, 成都市科龙化工试剂厂); N2气(纯度99.9%, 四川绵阳市昌俊气体有限公)
1.2实验方法
1.2.1溶液的配制铀标准溶液的配制:称取2.1092 g硝酸铀酰置于200 mL烧杯中, 加入适量超纯水搅拌至溶解后, 转入1000 mL棕色容量瓶中, 加入1.0 mL硝酸, 定容, 摇匀后即制得1000 mg/L的铀标准溶液. 不同浓度铀溶液分别用铀标准溶液稀释定容至所需浓度.
pH调节剂的配制:移取2.10 mL浓盐酸置于250 mL容量瓶中, 定容, 摇匀后即制得0.10 mol/L的盐酸标准溶液. 称取1.0 g氢氧化钠于100 mL烧杯中, 加入适量超纯水搅拌至溶解, 转入250 mL容量瓶中, 定容, 摇匀后即制得0.10 mol/L的氢氧化钠标准溶液.
U(Ⅵ) 测试显色剂的配制:称取0.1250 g偶氮胂Ⅲ于100 mL烧杯中, 加入适量超纯水搅拌至溶解, 转入500 mL棕色容量瓶中, 定容, 摇匀后得0.2 mmol/L的偶氮胂Ⅲ作为吸收光谱法测试U(Ⅵ)的显色剂.
实验基础溶液(U-NaCl体系)的配制:为增加实验溶液的导电性, 选择氯化钠作为添加电解质. 移取59.5 mL铀标准溶液置于500 mL容量瓶中, 称取1.4610 g氯化钠置于100 mL烧杯中, 加入适量超纯水搅拌至溶解, 移入上述含有硝酸铀酰的500 mL容量瓶中, 定容, 摇匀后得到含有1.0 mmol/L硝酸铀酰和0.10 mol/L氯化钠的实验基础液. 溶液pH=3.87, 放置7 d后溶液澄清无胶体沉淀.

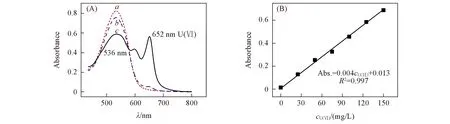
Fig.1 Absorbance spectra of ultra-pure water(a), 0.10 mol/L NaCl(b), 0.10 mol/L NaCl+100 mg/L U(Ⅵ)(c) complexing with arsenazo Ⅲ(A) and linear relationship between Abs. and cU(Ⅵ)(B)
1.2.3电化学表征电化学测试在传统的三电极体系电解池中进行. 选择氟掺杂SnO2导电玻璃(FTO)导电玻璃作为工作电极, 电极反应面积为15 mm×15 mm, 铂丝作为对电极, 饱和甘汞电极(SCE)作为参比电极, 利用PARSTAT4000电化学工作站(美国普林斯顿仪器公司)进行测试. 为减少电解液中溶解氧对电化学反应的干扰和影响, 在常规电化学测试之前向溶液中持续通入N2气(纯度99.9%)15 min[N2气对电化学还原U(Ⅵ)效率影响实验除外], 气体流量为50 mL/min.
采用循环伏安(CV)法对钠盐溶液中U(Ⅵ)的电子转移行为进行测试, 扫描电压范围为0.5~-1.2 V(vs. SCE), 扫描速度25 mV/s. 通过改变扫描速度(10, 25, 50, 75, 100, 250, 500, 750 mV/s), 探讨了U(Ⅵ)电子转移过程动力学特征. 利用pH调节剂, 调节实验基础溶液的pH值分别为1.5, 3.0, 4.5和6.0, 研究了pH值对U(Ⅵ)电子转移的影响.
根据U(Ⅵ)的还原电位, 在特定电位下进行恒电位沉积4 h, 间隔1 h取样. 采用交流阻抗谱图(EIS)及阻抗谱Zview拟合出的等效电路图, 在U(Ⅵ)的还原电位下, 对U(Ⅵ)晶化的反应动力学进行分析. 阻抗测试的频率为10000~0.1 Hz, 振幅为10 mV RMS(均值平方根).
1.2.4产物表征针对U(Ⅵ)在溶液中的络合形态, 采用地球化学计算软件MINTEQ进行模拟分析[21]. 反应前后工作电极经低温烘干, 表面物质的晶型结构采用X’Pert PRO X射线衍射仪(XRD, 荷兰帕纳科公司)测试表征. 2θ角扫描范围为3°~80°, 扫描速度为2°/min. 电极表面物质的形貌通过Ultra 55场发射扫描电子显微镜系统(SEM, 德国蔡司仪器公司)进行表征, 测试电压为15 kV, 物质元素成分由SEM自带的X射线能量色散谱仪(EDS, Oxford IE450X-Max80)测定.
2 结果与讨论
2.1U(Ⅵ)的电子转移过程
对NaCl溶液(0.10 mol/L, pH=3.87)和U-NaCl溶液体系[U(Ⅵ)浓度为1 mmol/L, pH=3.87]进行循环伏安扫描, 图2为在25 mV/s扫描速度下的循环伏安结果. 可看出, NaCl溶液的曲线无明显的氧化或还原峰出现. U-NaCl溶液体系在阴极扫描过程中出现了还原峰, 其中, 位于-0.25 V(vs. SCE)附近的还原峰B主要对应U(Ⅵ)还原为U(V)过程(反应3); 位于-0.50 V(vs. SCE)附近的还原峰C对应U(V)还原为U(Ⅳ)过程(反应4); 位于-1.10 V(vs. SCE)附近的还原峰D对应H+还原为H2过程(反应5). 在正向扫描过程中, 0.15 V(vs. SCE)附近出现的氧化峰A对应U(Ⅳ)氧化为U(Ⅵ)过程(反应6). 由测试结果可知, U(Ⅵ)还原成U(Ⅳ)分为两个过程, 而U(Ⅳ)一步氧化为U(Ⅵ). 这一现象与文献[18,19]结果相符. 已有研究结果表明, 在不同的实验体系条件下, U(Ⅵ)还原为U(Ⅳ)的过程是一个吸收能量的过程, 需要外部施加一定的条件, 整个还原过程分为两步; 而U(Ⅳ)氧化为U(Ⅵ)在自然条件下就可自发的进行且是一个快速的反应过程, 在施加一定的条件时, 表现为一步氧化[22].
(3)
(4)
(5)
(6)

Fig.2 Cyclic voltammogram of FTO electrode in 0.10 mol/L NaCl(solid line) and 1.0 mmol/L U(Ⅵ) + 0.10 mol/L NaCl(pH=3.87)(dotted line) at 25 mV/s
如图2所示, U(Ⅵ)与U(Ⅳ)之间转化存在可逆性. 为进一步探究U(Ⅵ)~U(Ⅳ)之间转化过程的动力学特征, 测试了U-NaCl体系在不同扫描速度下的循环伏安曲线. 由图3(A)可见, 氧化峰A和还原峰B(图3)的峰位并未随着扫描速度的增加而发生显著移动, 还原峰C随着扫描速度的增加而消失.
根据图3(A)中氧化峰A的峰电流值(jop)和还原峰B的峰电流值(-jrp)分别对扫描速度的1/2次方(ν1/2)作线性拟合[图3(B)], 可看出,jop和-jrp与ν1/2呈良好的线性关系, 与Randles Sevcik的控制扩散反应公式[式(7)]相符, 说明U(Ⅵ)的还原和氧化过程受扩散控制[23].
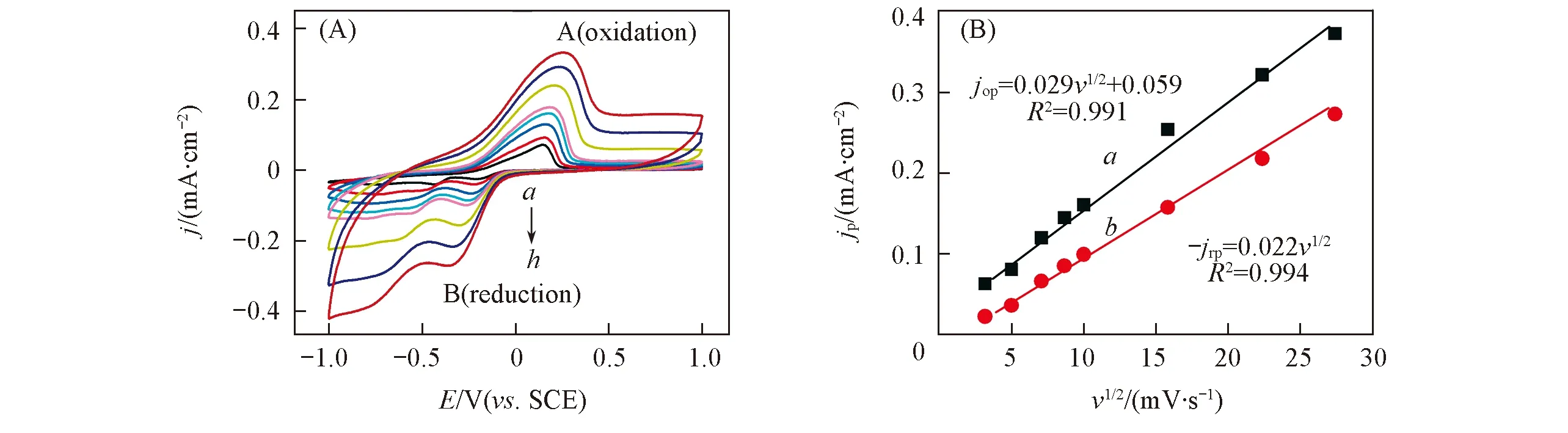
Fig.3 Cyclic voltammogram of FTO electrode in 1 mmol/L U(Ⅵ)+0.1 mol/L NaCl(pH=3.87) at diffe-rent scan rates(A) and linear relationship between jop, -jrp and ν1/2(B)(A) Scan rate/(mV·s-1):a. 10; b. 25; c. 50; d. 75; e. 100; f. 250; g. 500; h. 750. (B) a. for peak A(oxidation); b. for peak B(reduction).
(7)
随着扫描速度的增大, 还原峰C与还原峰B没有完全分开, 不能准确读取还原峰C的峰值, 可能是因为U(Ⅵ)得到电子后迅速还原为U(V), U(V)得到电子还原为U(Ⅳ)的同时发生歧化反应, U(V)迅速歧化为U(Ⅳ)和U(Ⅵ), 使U(V)得到电子转化为U(Ⅳ)的反应过程减弱, 扫描速度较大时, 不能明显地捕捉到U(V)转变为U(Ⅳ)的还原峰.
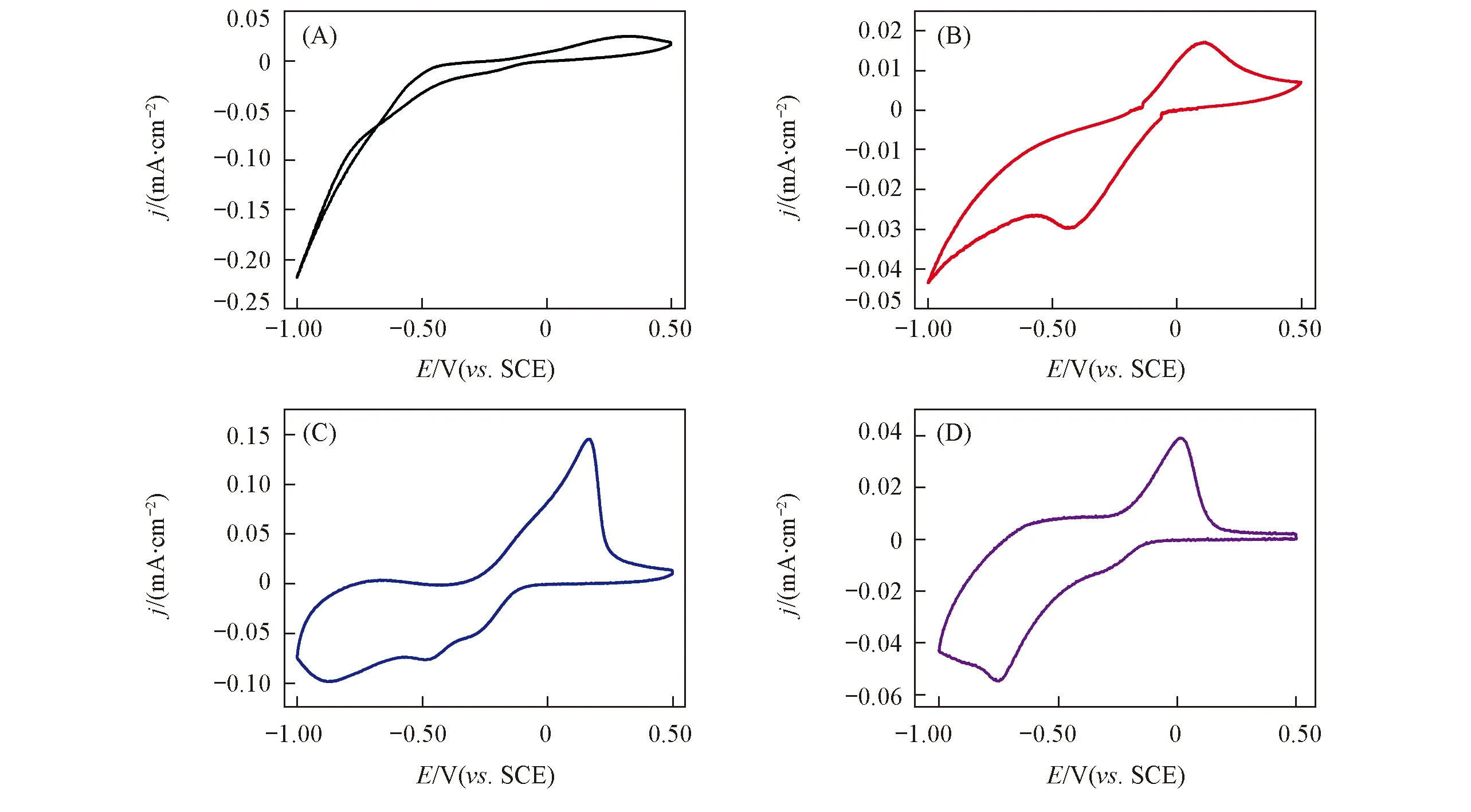
Fig.4 Cyclic voltammogram of FTO electrode in 1.0 mmol/L U(Ⅵ)+0.10 mol/L NaCl solution at pH values of 1.5(A), 3.0(B), 4.5(C) and 6.0(D) The scan rate was 25 mV/s.
2.2pH值对U(Ⅵ)电子转移过程的影响
由图4(A)可知, 在pH≤1.5的溶液中, U(Ⅵ)发生了快速的还原反应, 循环伏安曲线上无明显氧化还原峰; pH值增大到3.0[图4(B)]时曲线出现1个还原峰; 在pH值为4.5时出现2个还原峰[图4(C)], 回扫过程中均有氧化峰出现.
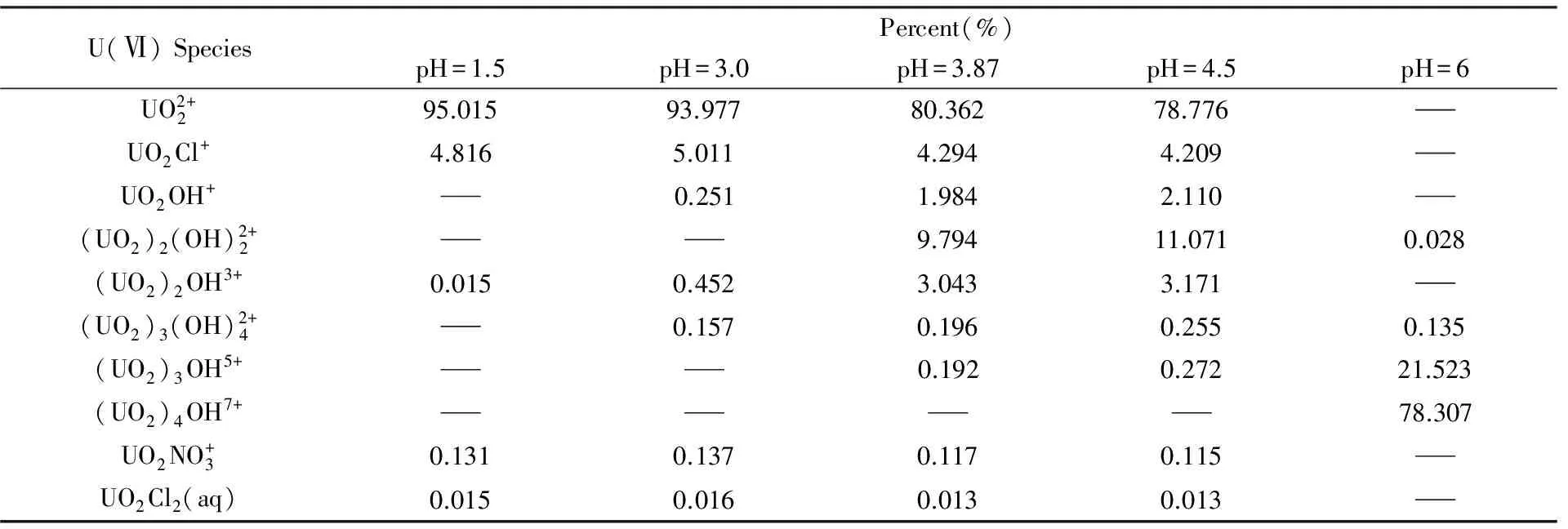
Table 1 Speciation of U(Ⅵ) at different pH conditions calculated by MINTEQ software*
*:“—” means the species does not exist.
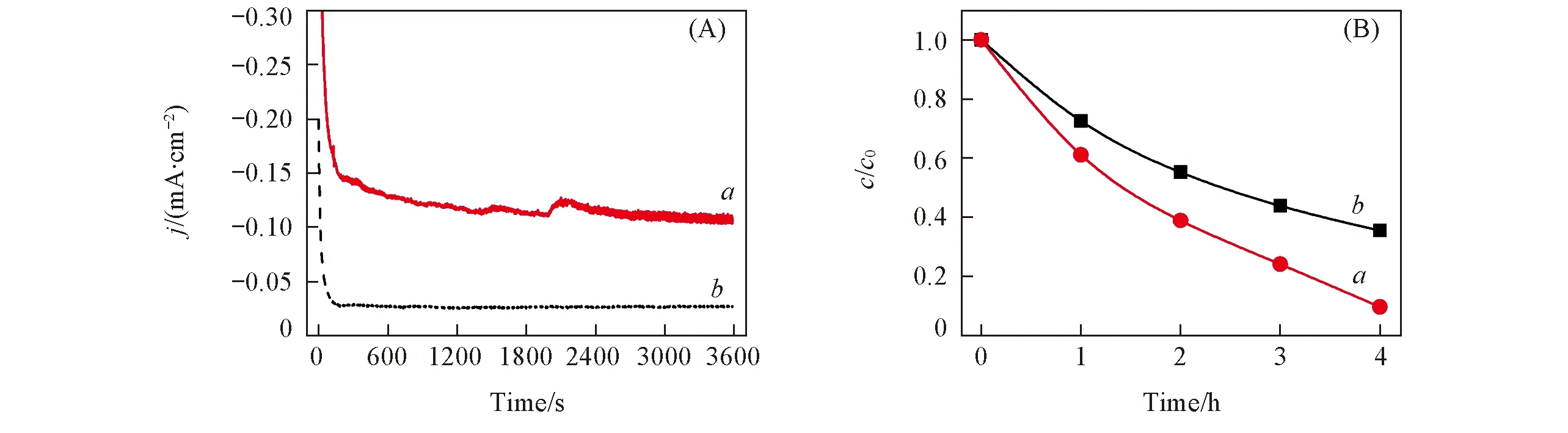
Fig.5 Amperometric i-t curve of FTO electrode in 1 mmol/L U(Ⅵ)+0.1 mol/L NaCl(pH=3.87) at a constant applied potential of -0.50 V(vs. SCE)(A) and variation of the U(Ⅵ) concentration with electrochemical reduction time(B) with(a) or without(b) N2-purging
2.3U(Ⅵ)的晶化研究
由图2的CV曲线测试结果可知, 当E≤-0.50 V(vs. SCE)时, U(Ⅵ)被电还原为U(Ⅳ), 因此选择在E=-0.50 V(vs. SCE)恒电位下研究电还原U(Ⅵ)的效果. 反应电流-时间曲线如图5(A)所示, 电极电流在200 s以后逐渐趋于稳定, 对应U(Ⅵ)离子在电极上的还原过程趋于稳定; 随着反应持续进行, 溶液中U(Ⅵ)浓度均逐渐降低[图5(B)]. 在持续通入氮气条件下, 经过4 h的电化学还原处理后, U(Ⅵ)被还原率达90%.
由图5还可知, 持续将N2气通入电解池时, 工作电极上的电流明显较大, U(Ⅵ)的还原效率也较高, 类似现象在光催化还原U(Ⅵ)的研究中也有出现[24]. 图6(A)为工作电极的交流阻抗谱图(EIS)及阻抗拟合图. 可见, EIS拟合曲线跟实验曲线基本一致, 拟合所得的等效电路图如图6(B)所示. 图中Rs代表电解液电阻,Rct代表电极反应过程中的电子转移电阻, CPE是常相位元件. 两种条件下等效电路图各个元件的拟合值如表2所示.

Fig.6 Electrochemical impedance spectra of FTO electrode in 1.0 mmol/L U(Ⅵ)+0.10 mol/L NaCl(pH=3.87) at a constant applied potential of -0.50 V(vs. SCE) with or without N2 purging(A) and equivalent circuit for FTO electrode(B)

SampleRs(Ω)/error(%)CPE1-T(F)/error(%)CPE1-P(F)/error(%)Rct(Ω)/error(%)WithoutN2-purging92.56/0.675.99×10-5/1.980.96/0.506340/6.02WithN2-purging84.56/0.987.10×10-5/2.940.94/0.754765/7.57
*Rs:Electrolyte resistance;Rct:charge transfer resistance; CPE:constant phase angle element. CPE1-T:Double layer capacitance values; CPE1-P:the contacting unevenness about polymer electrolyte of P electrode surface.
(8)
(9)
(10)
(11)
(12)
2.4晶化产物分析
随着反应时间的延长, 溶液中U(Ⅵ)的浓度[图5(B)]逐渐减小, 反应后电极表面有深色固体沉积[图7(A)]. 对比反应前后FTO电极表面的XRD表征结果[图7(B)]发现, 电极上的沉积产物主要为(UO2)6O2(OH)8·6H2O(PDF#:43-0364)、 UO2(PDF#:41-1422)和单质铀(PDF#:24-0749). 其中, (UO2)6O2(OH)8·6H2O(水铀矿)和UO2的衍射峰强度较强, 仅含有少量的单质铀.
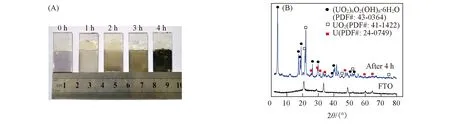
Fig.7 Photographs of FTO electrode after different time i-t measurements in 1.0 mmol/L U(Ⅵ)+0.10 mol/L NaCl(pH=3.87) at a constant applied potential of -0.50 V(vs. SCE) with N2-purging(A) and XRD pattern of FTO electrode before and after electrochemical reduction of U(Ⅵ) solution(B)
电极表面SEM照片[图8(A), (B)]显示, 电极表面产物主要为薄片状物质. 电子能谱[图8(C), (D)]结果显示, 电极表面产物的O/U平均原子比约为3.05, 而水铀矿(UO2)6O2(OH)8·6H2O 的O/U原子比为4.7, UO2的O/U原子比为2, 忽略少量单质铀不计, 经计算, 电极表面的沉淀物为38.9%水铀矿和61.1%的UO2, 主要还原产物为UO2. 电极表面沉淀物中存在水铀矿, 可能是因为在测试样品的前处理过程中, 还原后的产物暴露在空气中, 单质铀与空气中的氧发生反应生成UO2, 同时样品会与空气中的水分作用, 导致UO2发生变蚀进而生成水铀矿[25].

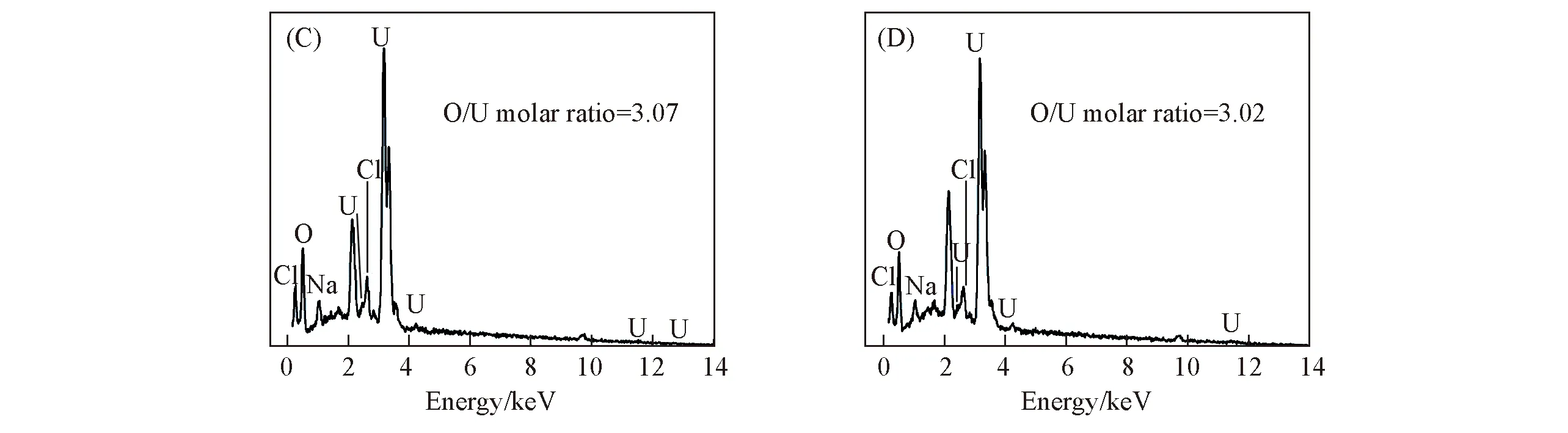
Fig.8 SEM images(A, B) and EDS spectra(C, D) of FTO electrode after electrochemical reduction in U(Ⅵ) solution for 4 h (C) and (D) are the EDS spectra of the framed area in (A) and (B), respectivery.
(13)
(14)
(15)
(16)
(17)
3 结 论
[1]Leggett R. W.,Health.Phys., 1989, 57(3), 365—383
[2]Domingo J. L.,Reprod.Toxicol., 2001, 15(6), 603—609
[3]Sharma P., Tomar R.,Micropor.Mesopor.Mater., 2008, 116(1/3), 641—652
[4]Xie S. B., Yang J., Chen C., Zhang X. J., Wang Q. L., Zhang C.,J.Environ.Radioactiv., 2008, 99(1), 126—133
[5]Xie S. B., Zhang C., Zhou X. H., Yang J., Zhang X. J., Wang J. S.,J.Environ.Radioactiv., 2009, 100(2), 162—166
[6]Ozay O., Ekici S., Aktas N., Sahiner N.,J.Environ.Manage, 2011, 92(12), 3121—3129
[7]Li H. Y., Wang B., Liu S. M.,Chem.J.ChineseUniversities, 2015, 36(4), 665—671(李宏宇, 王博, 刘树明. 高等学校化学学报, 2015, 36(4), 665—671)
[8]Chen L., Li Y., Wang Z. W., Peng Z. Y., Yang Z. M., Yuan L. H., Feng W.,Chem.J.ChineseUniversities, 2015, 36(8), 1485—1490(陈龙, 李艳, 王真文, 彭智勇, 杨泽明, 袁立华, 冯文. 高等学校化学学报, 2015, 36(8), 1485—1490)
[9]Abbasizadeh S., Keshtkar A. R., Mousavian M. A.,Biochem.Eng.J., 2013, 220(15), 161—171
[10]Sprynskyy M., Kovalchuk I., Buszewski B.,J.Hazard.Mater., 2010, 181(1—3), 700—707
[11]Wang G. H., Liu J. S., Wang X. G., Xie Z. Y., Deng N. S.,J.Hazard.Mater., 2009, 168(2/3), 1053—1058
[12]Wang G. H., Wang X. G., Chai X. J., Liu J. S., Deng N. S.,Appl.Clay.Sci., 2010, 47(3/4), 448—451
[13]Li X. Y., Zhang M., Liu Y. B., Li X., Liu Y. H., Hua R., He C. T.,Water.Qual.Expos.Hea, 2013, 5(1), 31—40
[14]Liang F. Y., Wu R. R., Cao C. L., Zheng Y., Yang C. H., Zhao F.,Chem.J.ChineseUniversities, 2014, 35(2), 372—376(梁方圆, 吴冉冉, 曹昌丽, 郑越, 杨朝晖, 赵峰. 高等学校化学学报, 2014, 35(2), 372—376)
[15]Wei Y. Z., Fang B., Arai T., Kumagai M.,J.Radioanal.Nucl.Ch., 2004, 262(2), 409—415
[16]Anderson C. J., Choppin G. R., Pruett D. J., Costa D., Smith W.,Radiochim.Acta, 1999, 84(1), 31—36
[17]Ikeda Y., Hiroe K., Asanuma N., Asanuma, N., Shirai A.,J.Nucl.Sci.Technol., 2009, 46(2), 158—162
[18]Zhang Q. Y., Huang X. H., Tang H. B., He H.,JournalofNuclearandRadiochemistry, 2011, 33(2), 101—105(张秋月, 黄小红, 唐洪彬, 何辉. 核化学与放射化学, 2011, 33(2), 101—105)
[19]Wang Y., Liu Y. P., Wang X. Y., Zhu T. W.,JournalofNuclearandRadiochemistry, 2013, 35(5), 263—269(王悦, 刘玉鹏, 王祥云, 褚泰伟. 核化学与放射化学, 2013, 35(5), 263—269)
[20]Tan X. F.,ResearchontheElectrochemicalBehaviorofUraniuminIonicLiquids, University of South China, Hengyang, 2014(谭绪凤. 铀在离子液体中的电化学行为研究, 衡阳:南华大学, 2014)
[21]Liu J. A., Feng X. G.,JournalofNuclearandRadiochemistry, 2011, 33(1), 32—41(刘杰安, 冯孝贵. 核化学与放射化学, 2011, 33(1), 32—41)
[22]Qiu L. Y., Yuan L. Y., Tan X. F., Shi W. Q., Liu L. J.,JournalofNuclearandRadiochemistry, 2014, 36(2), 65—74(邱凌云, 袁立永, 谭绪凤, 石伟群, 刘良军. 核化学与放射化学, 2014, 36(2), 65—74)
[23]Yuan K., Ilton E. S., Antonio M. R., Li Z., Cook P. J., Becker U.,Environ.Sci.Technol., 2015, 49(10), 6206—6213
[24]Kim Y. K., Lee S., Ryu. J., Park H.,Appl.Catal.B-Environ., 2015, 163(2), 584—590
[25]Ha D., Jin X.,YouKaangXuanYe, 1981, 4(3), 11—27(哈迪, 金鑫. 铀矿选冶, 1981, 4(3), 11—27)
(Ed.:S, Z, M)
† Supported by the State Key Development Program for Basic Research of China(No.2014CB846003), the National Natural Science Foundation of China(No.41272371) and the Fifty-nine Project was supported by the National Science Foundation for Post-doctoral Scientists of China(No.2016M592698).
Electrochemical Electron Transfer and Crystallization Process of Uranium(Ⅵ) in Sodium Salt Solution†
ZONG Meirong1, HE Huichao2, DONG Faqin1, 2*, HE Ping2, SUN Shiyong1,LIU Mingxue1, NIE Xiaoqin1
(1. Key Laboratory of Solid Waste Treatment and Resource Recycle, Ministry of Education,SouthewestUniversityofScienceandTechnology,Mianyang621010,China;2.StateKeyLaboratoryCultivationBaseforNonmetalCompositesandFunctionalMaterials,SouthewestUniversityofScienceandTechnology,Mianyang621010,China)
Electrochemical beheaviors of uranium(VI) was investigated with cyclic voltammetry and chronoamperometry(CV). The reduction of U(Ⅵ) was analyzed by potentiostatic electrochemical and the kinetics was investigated with electrochemical impedance spectroscopy(EIS). In addition, crystallization of U(Ⅵ) were analyzed by X-ray diffraction(XRD), scanning electron microscopy(SEM) and energy-dispersive spectrometry(EDS). The results indicated that the reduction processes of U(Ⅵ) to U(Ⅳ) was significantly in fluenced by the pH value of solution, with the pH value changed from 3.87 to 4.50, the cyclic voltammetry showed that the reduction of U(Ⅵ) to U(Ⅳ) was in a manner of two-step one-electron process with a diffusion-controlled reaction mechanism. The cyclic voltammetry results presented a constant potential reduction method for removing U(Ⅵ) from aqueous solution, and the reduction efficiency can reach 90%. At a constant potential, U(Ⅵ) mainly crystalized into solid phases in the forms of UO2and (UO2)6O2(OH)8·6H2O. These results could provide a basis for removing and collecting U(Ⅵ) from solution.
Uranium(Ⅵ); Electrochemistry; Electrochemical impedance spectroscopy; Crystallization
10.7503/cjcu20160171
2016-03-23. 网络出版日期:2016-08-26.
国家“九七三”计划项目(批准号:2014CB846003)、 国家自然科学基金(批准号:41272371)和中国博士后科学基金第59批面上基金(编号:2016M592698)资助.
O646
A
联系人简介:董发勤, 男, 博士, 教授, 主要从事环境友好材料和地球科学方面的研究. E-mail:fqdong@swuat.edu.cn
