牦牛发情期卵巢比较转录组学研究
2016-11-01兰道亮熊显荣柴志欣
兰道亮,熊显荣,柴志欣,艾 鷖,黄 偲,李 键*
(1.西南民族大学 青藏高原研究院,成都 610041;2.西南民族大学 生命科学与技术学院,成都 610041)
牦牛发情期卵巢比较转录组学研究
兰道亮1,2,熊显荣2,柴志欣1,艾鷖1,黄偲2,李键1,2*
(1.西南民族大学 青藏高原研究院,成都 610041;2.西南民族大学 生命科学与技术学院,成都 610041)
旨在进一步了解牦牛发情期卵巢的分子机制,解析牦牛繁殖的特殊性。本研究应用RNA-seq技术对牦牛和平原黄牛发情期卵巢进行转录组高通量测序及全基因组差异表达模式比对分析。通过比较分析牦牛和黄牛卵巢转录组数据,共筛选出1 307个差异表达基因,其中661个基因表达量上调和646个基因表达量下调。进一步功能分析表明,这些差异基因涉及多种 GO分类及KEGG通路。其中GO分类注释显示,差异基因与细胞粘附、激素调控等生物学过程存在密切关联,同时钙离子结合、阳离子跨膜转运等分子事件表现活跃。KEGG通路分析显示,补体和凝血级联通路的富集水平最高,其次为细胞色素P450相关通路。昼夜节律等一些新型通路,尽管与生殖功能没有明显关联,但也表现出显著富集。本研究首次对比牦牛和黄牛发情期卵巢转录组数据,筛选并分析相关差异基因。该研究结果为进一步阐述牦牛卵巢的基本分子机理提供基础,同时也为全面理解牦牛繁殖特异性提供新的思路。
牦牛;发情期卵巢;转录组;比较分析;分子机制
牦牛(Bosgrunniens)素有 “高原之舟”的美誉,是主要分布在中国青藏高原及其毗邻高山或亚高山地区的特有物种,也是当今世界上已知唯一一种能够在高海拔地区(平均海拔高度3 000米)生存的牛属动物[1-2]。牦牛能够很好地适应高山草原环境,并在恶劣的高原环境条件下(如空气稀薄、低温和草料短缺)自由生长和繁殖[3]。同时牦牛可以为当地牧民提供奶、肉、毛等丰富的生产和生活资料,是高原地区不可或缺的重要畜种[1,4-5]。与生活在平原地区的普通牛类相比,牦牛的性成熟发育得更慢,其生殖能力普遍更低[6-7]。一头成年牦牛的平均繁殖率仅为48.61%,其中超过一半的牦牛为每两年生育一胎或每三年生育两胎。此外,雌性牦牛发情率也很低,超过90%产后雌性牦牛无法在同年的发情期发情[8-9]。
卵巢是雌性哺乳动物的重要生殖器官。它具有众多功能,包括提供能受精的卵母细胞、分泌生殖激素以及维持雌性动物发情周期。卵巢功能直接影响着雌性动物的繁殖能力[10]。每个发情周期过程中,卵巢都会经历增殖、侵袭、分化和细胞凋亡;这些正常的生理变化直接影响和决定了雌性动物的排卵、受精率和产仔数[11]。与其他普通牛类相比,牦牛的卵巢较小,卵巢系膜更短,其位置也相对固定,但与前者的整体结构是相似的[12]。迄今为止,针对牦牛卵巢的研究多集中于其形状和解剖结构,而对牦牛卵巢的分子基础及其分子机制仍然尚不明确。卵巢功能的实现是一个复杂的过程,这个过程涉及大量基因的转录调控。另外,基因表达的差异性是物种进化的重要组成部分,也是产生生物多样的根本途径[11,13]。因此,为了解牦牛卵巢的分子机制和牦牛繁殖的特殊性,需要对其转录组学进行研究。随着高通量测序技术的发展,RNA高通量测序(RNA sequencing)为大范围的转录学研究提供强有力的工具,与传统方法相比,其优势十分明显[11]。而且随着牦牛基因组测序的完成也使进一步的转录组学研究成为可能[1]。本研究应用RNA-seq技术对牦牛和平原黄牛发情期卵巢进行转录组学测序及比对分析,深入了解牦牛卵巢在发情周期中的差异表达基因及其调控通路,为进一步研究牦牛卵巢的分子机制及全面了解牦牛繁殖的特异性提供基础。
1 材料与方法
1.1样本采集
参照文献[14-15]的方法对牦牛的发情期进行鉴定。从高原屠宰场(中国四川红原,纬度31°51′ N~33°19′ N,经度101°51′ E~103°23′ E;平均海拔高度为3 600 m)随机选择3头处于自然发情周期且体型相当的健康4岁雌性麦洼牦牛。同样从高原屠宰场(中国四川成都,纬度30°05′ N~31°26′ N,经度102°54′ E~104°53′ E;平均海拔高度为750米)随机选择3头处于自然发情期的健康4岁雌性黄牛(Bostaurusdomesticus)。屠宰后立即采集牦牛和黄牛的卵巢组织并进行观察确认后,将组织放液氮中保存备用。
1.2RNA提取、cDNA文库构建和Illumina测序
应用TRIzol试剂(Life Technologies公司,美国)提取牦牛和黄牛卵巢组织的总RNA。提取后的各RNA样品均以无RNase的DNase I(TaKaRa公司,大连)进行处理,以消除可能存在基因组DNA污染。然后分别等量混合3个牦牛和黄牛的RNA样品,组成两个RNA池(RNA Pool,各30 μg)。使用Oligotex mRNA小量提取试剂盒(Qiagen公司,德国)从RNA池中分离和纯化mRNA。整个流程 RNA 的质量和含量通过Agilent 2100生物分析仪(Agilent公司,美国)进行测定。使用 TruSeq RNA Sample Prep Kit试剂盒(Illumina公司,美国),参照其说明书标准流程构建随机片段测序文库。即用中段试剂在恒温混匀仪中对纯化的mRNA进行片段化(约200 bp),而后将其作为cDNA第一链合成的模板。随后使用RNase H、dNTP及DNA聚合酶I合成第二链cDNA。纯化和配对末端修复后,将cDNA片段连接到测序接头并通过聚合酶链反应(PCR)对其进行扩增,以此获得最终的双末端(Paired-end,PE)测序文库。使用Agilent2100生物分析仪和ABI StepOnePlus实时PCR系统执行质控(QC)测试后,在Illumina HiSeq 2500平台上对文库进行测序。
1.3转录组数据分析
对Hiseq 2500测序产生的原始读数进行QC测试后,通过去除接头序列、空序列及低质量测序序列(≤Q20质量值),将原始读数过滤为净测序序列(Clean reads)。使用SOAPaligner/SOAP2软件,将过滤后序列与牦牛基因组(版本1.0)进行比对[16]。然后利用比对数据计算这些测序序列在基因组及基因上的分布,并统计覆盖率。根据RPKM法 (Reads per kilobase transcriptome per million mapped reads)计算基因表达水平。采用 “数字基因表达谱显著性”算法(The significance of digital gene expression profiles)界定牦牛和普通牛类文库之间的差异表达基因(Differential expressed genes,DEGs)[17],同时用P值检测对应达到统计学显著水平的差异基因表达。伪发现率(FDR)≤0.001和log2Ratio的绝对值≥1作为确定基因表达差异显著性的阈值。通过GO和KEGG通路富集分析(P≤0.05),对DEGs的功能进行注释。
1.4Real-time PCR (qRT-PCR) 验证
从测序的差异表达基因中随机选择15个基因进行qRT-PCR验证(基因、引物信息见附表1)。使用TRIzol试剂(Life Technologies公司,美国)提取同期取样的牦牛和黄牛卵巢组织总RNA。用无核酸酶ddH2O将各RNA样品的浓度调整至1 μg·μL-1,并用SuperScript®Ⅲ第一链合成系统(Life Technologies公司,美国)在20 μL的反应系统中对2 μg的总RNA进行反转录。然后使用SYBRH Premix Ex TaqTM Ⅱ(Tli RNaseH Plus)试剂盒(TaKaRa公司,大连)和Bio-Rad CFX96 TouchTM实时PCR系统(Bio-Rad公司,美国)进行qRT-PCR操作。反应条件:95 ℃ 1 min,95 ℃ 10 s,60 ℃ 40 s,总共40个循环。反应结束后进行最终熔解度曲线分析。采用β-Actin作为内参基因,根据2-ΔΔCt法计算各基因的相对表达量。
2 结 果
2.1转录组测序数据分析
转录组测序数据已递交于NCB数据库(接受号:SRX335453、SRX1302580)。去除低质量序列(即仅包含接头、接头序列的读数和空读序列)后,在牦牛转录测序文库中共获得53 653 032条过滤测序序列,包含4 828 772 880 碱基(bp)。在黄牛转录测序文库中共获得54 855 396条过滤测序序列,包含4 936 985 640碱基(bp)。碱基组成和质量分析结果显示,原始测序数据表现出均衡的碱基组成,且牦牛和普通黄牛文库的Q20(即碱基质量值≥20)的比率分别为94.7%和94.5%,这一结果表明,文库构建成功且测序质量良好。本研究以牦牛基因组作为参考基因组。比对分析显示,牦牛文库中有33 168 751条(61.82%)测序序列、普通黄牛文库中有31 730 973条测序序列(57.84%)可比对至牦牛基因组。基因覆盖率统计表明,牦牛和普通黄牛发情期卵巢文库中分别有16 992条和16 904条牦牛基因得到映射。此外,笔者发现一定比例的、无法映射至现有基因的测序序列(约30%),但是这些测序序列均位于基因组上,为进一步挖掘牦牛新转录子或基因提供数据基础。
2.2差异表达基因筛选
通过比较分析牦牛和黄牛发情期卵巢转录组数据,共筛选出1 307个差异表达基因,其中661个基因表达量上调和646个基因表达量下调。为验证这些基因表达数据,笔者随机选择15个差异表达基因,以同期取样的牦牛和黄牛卵巢组织为样本进行qRT-PCR检测。结果显示,这些差异基因的基本表达模式表现出与转录组数据相似的变化趋势(表1),表明转录组测序结果可靠。
2.3差异基因功能分析
为深入了解差异表达基因的生物学意义,笔者对这些差异表达基因进行GO和KEGG富集分析。GO富集分析表明,差异表达基因在分布于生物学过程(Biological processes,BP)、细胞组分(Cellular components,CC) 及 分 子 功 能 (Molecular functions,MF)3大类中的GO类别上均出现富集。按生物过程分类,共有3 067个GO类别出现富集,富集前10的类别(Top 10)已列于表1中。在富集前10的GO类别中,“生物粘附”(Biological adhesion)、“细胞粘附”(Cell adhesion)和“细胞间粘附”(Cell-cell adhesion)等3个粘附相关GO类别的富集水平最高。此外,与激素相关的两个GO类别,即“类固醇激素刺激响应”(Response to steroid hormone stimulus)和“雌激素刺激响应”(Response to estrogen stimulus),也富集于前10 GO类别中。在分子功能的分类下,共有667个GO类别出现富集,位列前10的显著富集GO类别已列于表2中。在位列前10的GO类别中,“钙离子结合”(Calcium ion binding)的富集水平最高。在前10富集GO类别中,阳离子跨膜转运相关(Cation transmembrane transport-related)GO类别占据了绝大部分(6/10)。在细胞组分的分类下,共有427个GO类别出现富集。在位列前10的富集GO类别中(表3),所有类别均属于细胞外(Extracellular parts)或质膜部分(Plasma membrane parts)。KEGG富集分析表明,DEGs中共有243个通路出现富集,在位列前10的通路中(表4),“补体和凝血级联”(Complement and coagulation cascades)通路的富集水平最高,其次 “细胞色素P450引起的外源性物质代谢”(Metabolism of xenobiotics by cytochrome P450)和“药物代谢-细胞色素P450”(Drug metabolism-cytochrome P450)等与细胞色素P450相关通路也得到显著富集。一些与卵巢生殖功能无显著关联的新型通路如“昼夜节律”通路(Circadian rhythm)也出现在富集水平前10的通路中。
表1生物学过程前10富集 GO 分类
Table 1Top 10 enrichment GO categories in the biological processes of DGEs
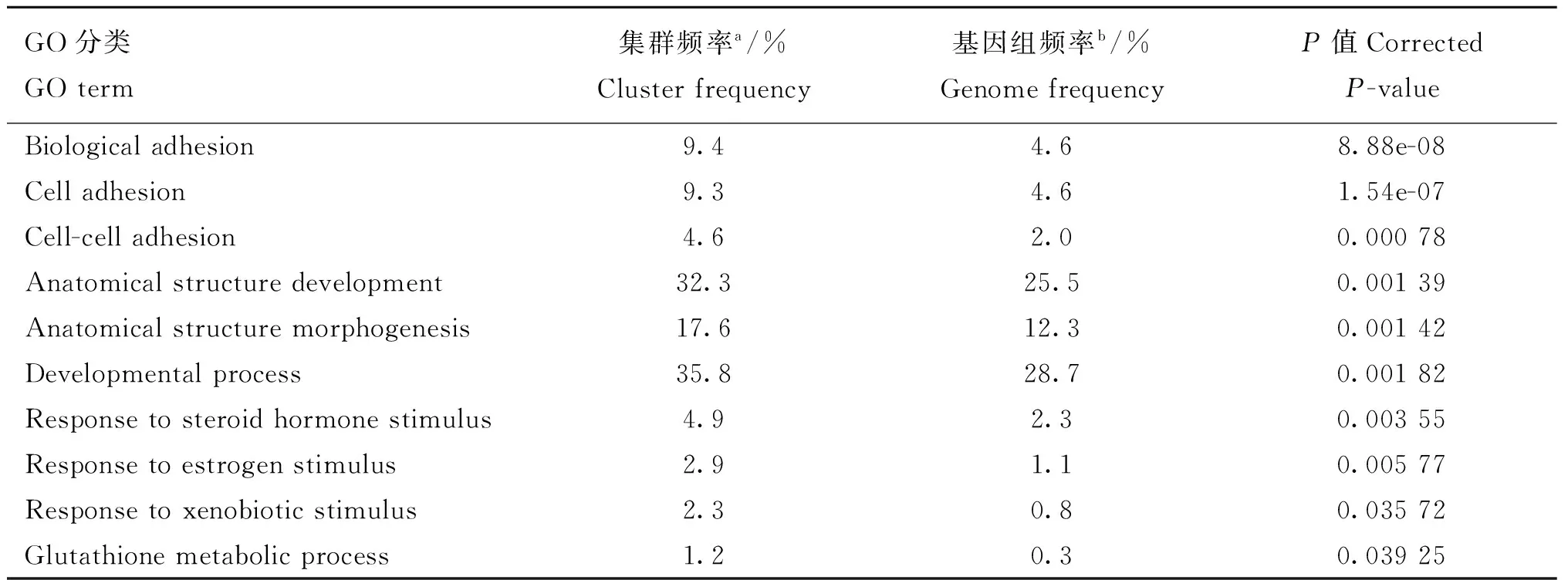
GO分类GOterm集群频率a/%Clusterfrequency基因组频率b/%GenomefrequencyP值CorrectedP-valueBiologicaladhesion9.44.68.88e-08Celladhesion9.34.61.54e-07Cell-celladhesion4.62.00.00078Anatomicalstructuredevelopment32.325.50.00139Anatomicalstructuremorphogenesis17.612.30.00142Developmentalprocess35.828.70.00182Responsetosteroidhormonestimulus4.92.30.00355Responsetoestrogenstimulus2.91.10.00577Responsetoxenobioticstimulus2.30.80.03572Glutathionemetabolicprocess1.20.30.03925
a.不同GO分类中的差异基因占所有GO分类总差异基因(965)的比例;b.该GO分类包含的基因占基因组所有基因(15 573)的比例
a.The proportion of differentially expressed genes with biological processes GO categories in total differentially expressed genes(965)with GO annotation;b.The proportion of genes with GO annotation in the entire yak genome(15 573)
表2分子功能过程前10富集 GO 分类
Table 2Top 10 enrichment GO categories in the molecular functions of DGEs
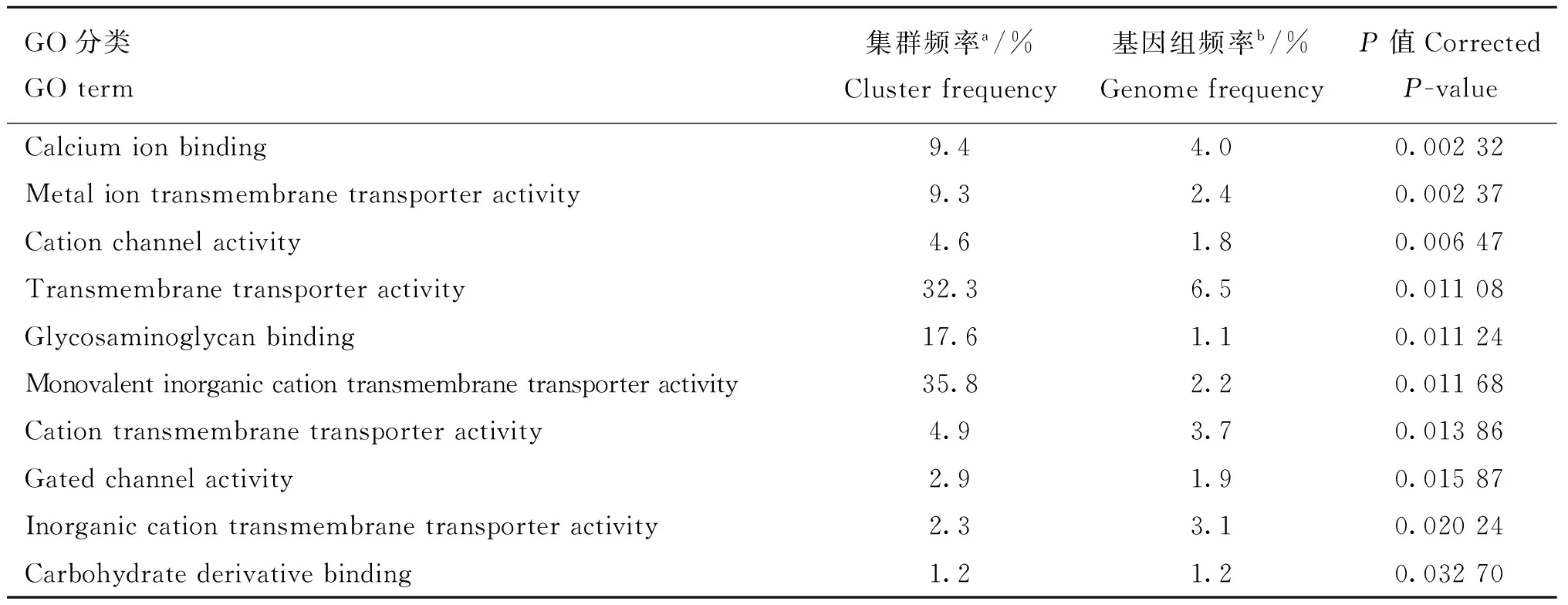
GO分类GOterm集群频率a/%Clusterfrequency基因组频率b/%GenomefrequencyP值CorrectedP-valueCalciumionbinding9.44.00.00232Metaliontransmembranetransporteractivity9.32.40.00237Cationchannelactivity4.61.80.00647Transmembranetransporteractivity32.36.50.01108Glycosaminoglycanbinding17.61.10.01124Monovalentinorganiccationtransmembranetransporteractivity35.82.20.01168Cationtransmembranetransporteractivity4.93.70.01386Gatedchannelactivity2.91.90.01587Inorganiccationtransmembranetransporteractivity2.33.10.02024Carbohydratederivativebinding1.21.20.03270
a.不同GO分类中的差异基因占所有GO分类总差异基因(944)的比例;b.该GO分类包含的基因占基因组所有基因(15 811)的比例
a.The proportion of differentially expressed genes with molecular functions GO categories in total differentially expressed genes(944)with GO annotation;b.The proportion of genes with GO annotation in the entire yak genome(15 811)
表3细胞组分过程前10富集 GO 分类
Table 3Top 10 enrichment GO categories in the cellular components of DGEs
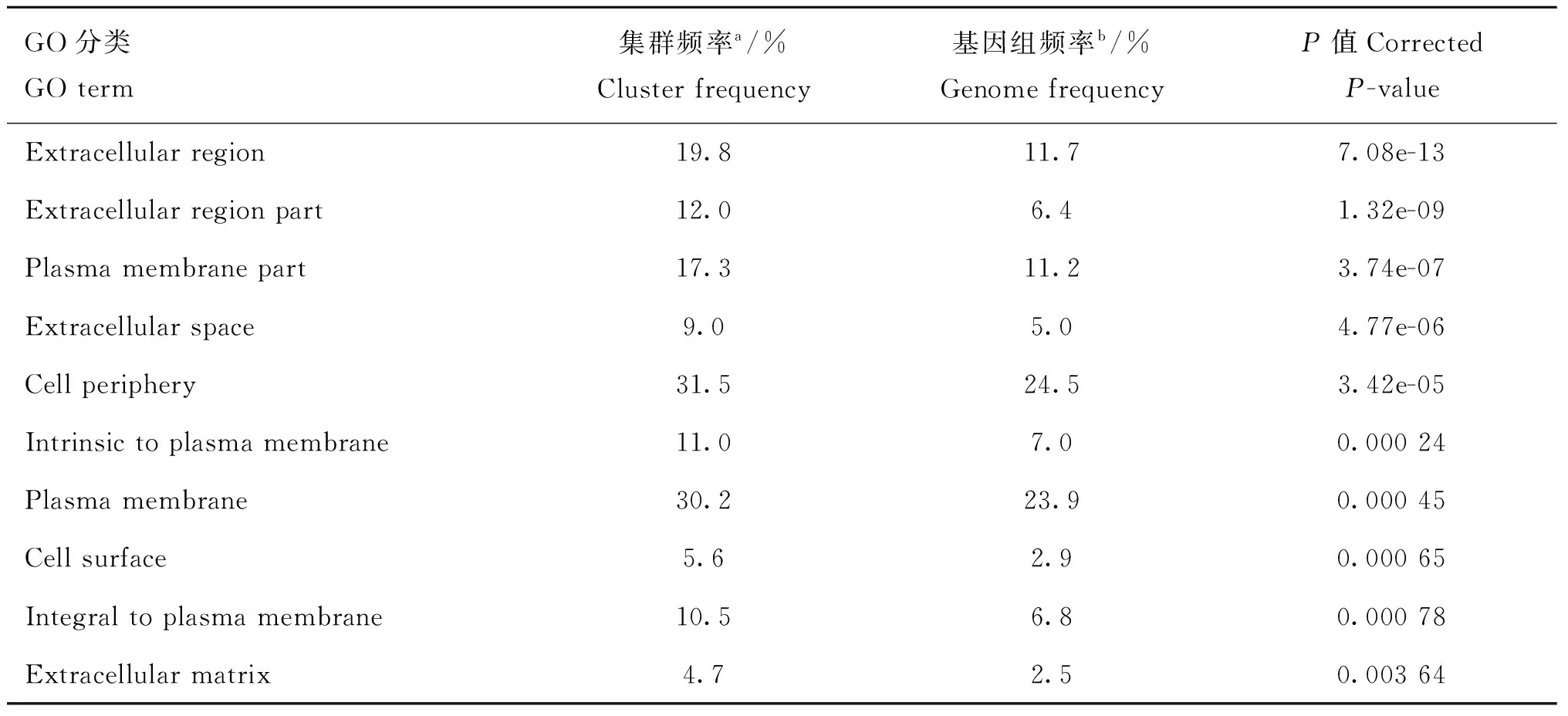
GO分类GOterm集群频率a/%Clusterfrequency基因组频率b/%GenomefrequencyP值CorrectedP-valueExtracellularregion19.811.77.08e-13Extracellularregionpart12.06.41.32e-09Plasmamembranepart17.311.23.74e-07Extracellularspace9.05.04.77e-06Cellperiphery31.524.53.42e-05Intrinsictoplasmamembrane11.07.00.00024Plasmamembrane30.223.90.00045Cellsurface5.62.90.00065Integraltoplasmamembrane10.56.80.00078Extracellularmatrix4.72.50.00364
a.不同GO分类中的差异基因占所有GO分类总差异基因(1 054)的比例;b.该GO分类包含的基因占基因组所有基因(17 161)的比例
a.The proportion of differentially expressed genes with cellular components GO categories in total differentially expressed genes(1 054)with GO annotation;b.The proportion of genes with GO annotation in the entire yak genome(17 161)
表4富集前 10 (Top 10) KEGG 通路列表
Table 4Top 10 enrichment KEGG pathways in the DGEs
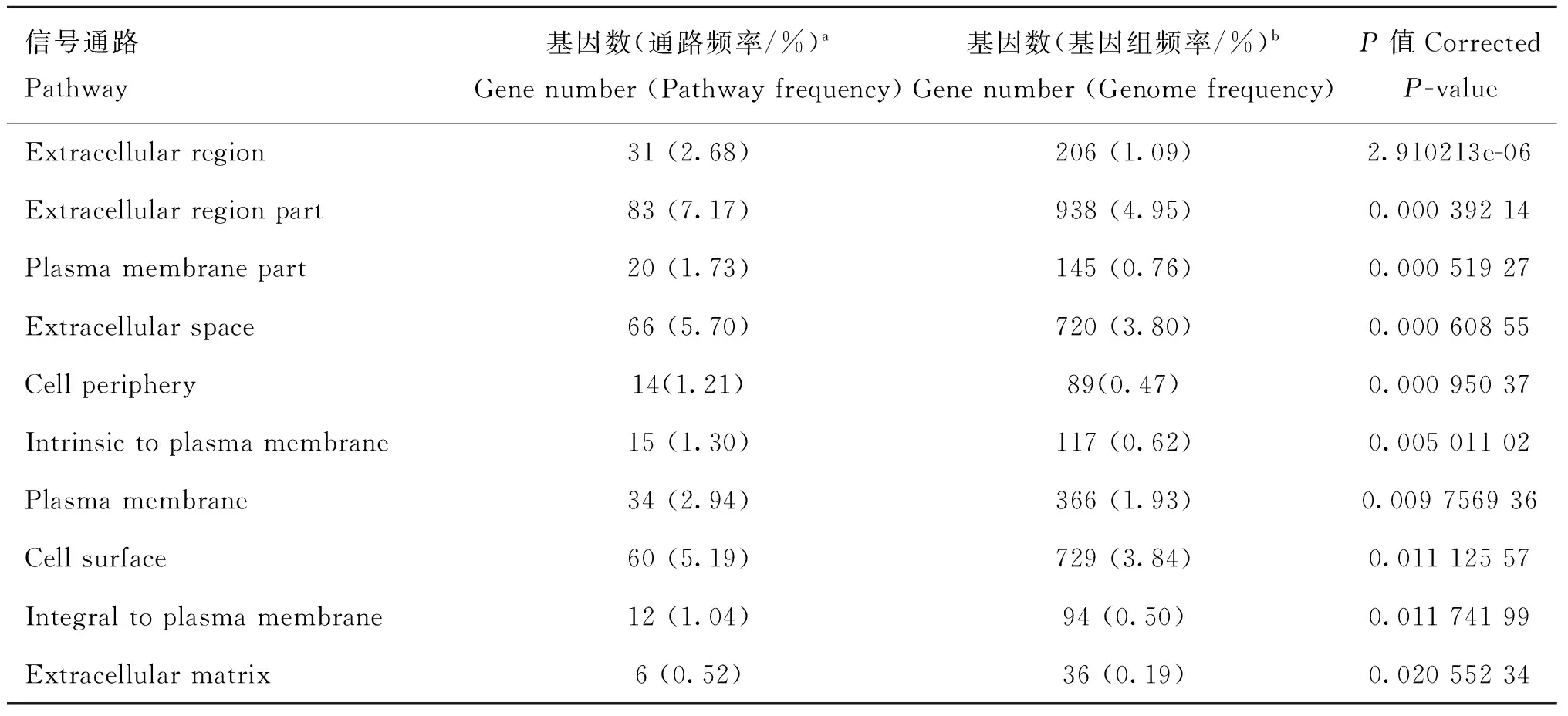
信号通路Pathway基因数(通路频率/%)aGenenumber(Pathwayfrequency)基因数(基因组频率/%)bGenenumber(Genomefrequency)P值CorrectedP-valueExtracellularregion31(2.68)206(1.09)2.910213e-06Extracellularregionpart83(7.17)938(4.95)0.00039214Plasmamembranepart20(1.73)145(0.76)0.00051927Extracellularspace66(5.70)720(3.80)0.00060855Cellperiphery14(1.21)89(0.47)0.00095037Intrinsictoplasmamembrane15(1.30)117(0.62)0.00501102Plasmamembrane34(2.94)366(1.93)0.009756936Cellsurface60(5.19)729(3.84)0.01112557Integraltoplasmamembrane12(1.04)94(0.50)0.01174199Extracellularmatrix6(0.52)36(0.19)0.02055234
a.不同通路中的差异基因占所有通路总差异基因(1 157)的比例;b.该通路包含的基因占基因组所有基因(18 965)的比例
a.The proportion of differentially expressed genes with pathway annotation in total differentially expressed genes(1 157)with pathway annotation;b.The proportion of genes with GO annotation in the entire yak genome(18 965)
3 讨 论
为了解牦牛卵巢的分子机制和牦牛繁殖的特殊性,本研究应用RNA-seq技术对牦牛和平原黄牛发情期卵巢进行转录组学测序及比对分析,共筛选获得1 307个差异表达基因,进一步功能分析这些差异表达基因涉及多个GO分类和KEGG信号通路。在生物过程GO分类中,发现“生物粘附”(Biological adhesion)、“细胞粘附”(Cell adhesion)和“细胞间粘附”(Cell-cell adhesion)等3个粘附相关GO类别的富集水平最高。以往的研究表明,卵巢中的卵泡处于无血管的微环境中,卵母细胞、卵丘细胞和其他卵泡细胞间存在广泛的间隙连接,从而形成一个功能完备的联合体。卵母细胞主要通过细胞黏附和连接与其周围细胞(卵泡颗粒及膜细胞)进行通讯[18-19]。本研究结果表明,处于发情周期中的牦牛和黄牛卵巢间可能存在不同的细胞粘附方式。在分子功能GO分类中,在前10富集的GO类别中,“钙离子结合”(Calcium ion binding)的富集水平最高。作为第二细胞内信使,钙离子调节着细胞中许多重要的生理和病理过程。除了调节哺乳动物卵母细胞的增长和减数分裂、参与激素和生长因子分泌外,钙离子也与卵母细胞的减数分裂成熟调节密切相关[20-21]。“钙离子结合”类别在本研究中的富集水平最高,这说明牦牛和黄牛卵巢间的卵母细胞减数分裂成熟调节可能存在差异。在前10富集GO类别中,阳离子跨膜转运相关(Cation transmembrane transport-related)GO类别占据大部分(6/10)。细胞膜的离子活性与细胞内环境调节密切相关,如pH、渗透压、营养吸收和膜电位;它在神经递质分泌和激素代谢调节中也起着重要的作用[22]。另外,离子跨膜转运建立的细胞膜浓度梯度为一系列细胞代谢活动提供能量来源。在许多阳离子跨膜转运活动中,钙和锌离子的转运在卵母细胞成熟调节中发挥着重要作用[23-25]。本研究中,阳离子跨膜转运相关GO类别的富集证实,与黄牛类卵巢相比,牦牛卵巢的离子跨膜转运调节有所不同,但其确切功能仍有待进一步研究。在细胞组分的分类下,位列前10的富集GO类别中,所有类别均属于细胞外或质膜部分。这一结果与生物过程和分子功能分类一致。例如,粘附、激素和阳离子跨膜转运相关类别均发生于细胞外或质膜部分,进一步证实富集的准确性。
在KEGG富集分析中,其中“补体和凝血级联”(Complement and coagulation cascades)通路的富集水平最高。补体是在具有酶活性组织液中发现的一组不耐热性球蛋白,在先天性和获得性免疫反应中发挥着重要作用[26]。补体通过经典、凝集素和旁路这3种途径,可以激活一系列级联反应,进而触发补体分子效应。激活后的补体可以调节吞噬细胞和免疫细胞,并增强炎症反应;还可以通过溶解细胞壁直接杀灭某些细菌或细胞[27]。血凝是指血浆中的纤维蛋白通过外在和内在凝血级联通路转化为不溶性纤维蛋白的过程[28]。“补体和凝血级联”通路属于一种内源性代谢级联。除了主体生理功能,该通路也与许多其他生理和病理调节过程密切相关。例如,补体系统在许多生物过程中都起着重要作用,其中包括繁殖、发育、干细胞分化和组织再生[29-30]。补体系统的Bf和DAF因子在卵细胞和配子的卵泡发育和成熟中扮演重要角色[31]。如图1所示,3种级联方式激活补体的关键基因的表达水平都出现升高。这一结果说明,包括细胞裂解、肌肉收缩、趋化、吞噬细胞复原和炎症在内的下游生理功能可能得到增强。在凝血级联通路中发现,激活凝血的外在通路中基因仅出现少量表达。表明,包括血管损伤、纤维蛋白降解产物和一氧化氮生物合成在内的主要生理功能可能出现下降。然而,内在通路中的基因也出现大量表达,表明包括炎症和细胞粘附在内的相关下游生理功能可能被加强。导致细胞粘附过程的强化与GO类别生物过程的富集结果是相一致的,证明细胞粘附过程是牦牛和黄牛卵巢生理活动的重要差异之一。该结果表明,“补体和凝血级联”通路在牦牛卵巢的生理活动中具有重要作用,除能够调节免疫力、炎症外还可能与繁殖和发育密切相关。该通路可能与牦牛生活的特殊环境有关。为了抵御恶劣的高原环境引起的卵巢损伤和功能障碍,牦牛可能形成一种比黄牛更强的分子防御机制。
在位列前10的KEGG通路中,“细胞色素P450引起的外源性物质代谢” (Metabolism of xenobiotics by cytochrome P450)和“药物代谢-细胞色素P450”(Drug metabolism-cytochrome P450)等与细胞色素P450相关通路也出现富集。细胞色素P450为一类亚铁血红素-硫醇盐蛋白的超家族,主要包括CYP1、CYP2和CYP3亚家族。细胞色素P450是一种重要的代谢酶系统,可以催化体内多种内源性和外源性化合物的氧化反应,更改化学物质中的活性基团或有毒化合物,以及改变药物的疗效。细胞色素P450参与外源物质的生物转化,并与内源性物质的代谢存在关联,同时它也是机体代谢调节中一种重要的化合物[32]。此外,细胞色素P450参与了固醇和类固醇激素的生物合成,尤其是性激素的生物合成,在生殖激素调节中发挥着重要的作用[33-34]。这一结果与生物过程中激素相关GO类别的富集相一致,证明牦牛和黄牛卵巢之间存在着激素调节系统方面的差异。缺氧,包括高海拔缺氧,能够显著改变各细胞色素P450亚家族中的基因表达水平。缺氧诱导因子1(HIF-1),一种在低氧刺激下调节耐缺氧基因的重要因子,尤其是在高海拔缺氧调节扮演重要角色。研究表明作为细胞色素P450成员的CYP3A6启动子可以与HIF-1基因的特异性和转录激活相结合,从而诱导耐低氧基因的表达[35]。细胞色素P450的成员CYP17A1和CYP2E1基因也与藏族人存在密切联系,能够帮助他们适应高原的低氧环境[36]。牦牛长久以来一直生活在青藏高原。鉴于本研究中有两个细胞色素P450通路出现显著富集,可能也与牦牛对高原低氧环境的适应能力有关。
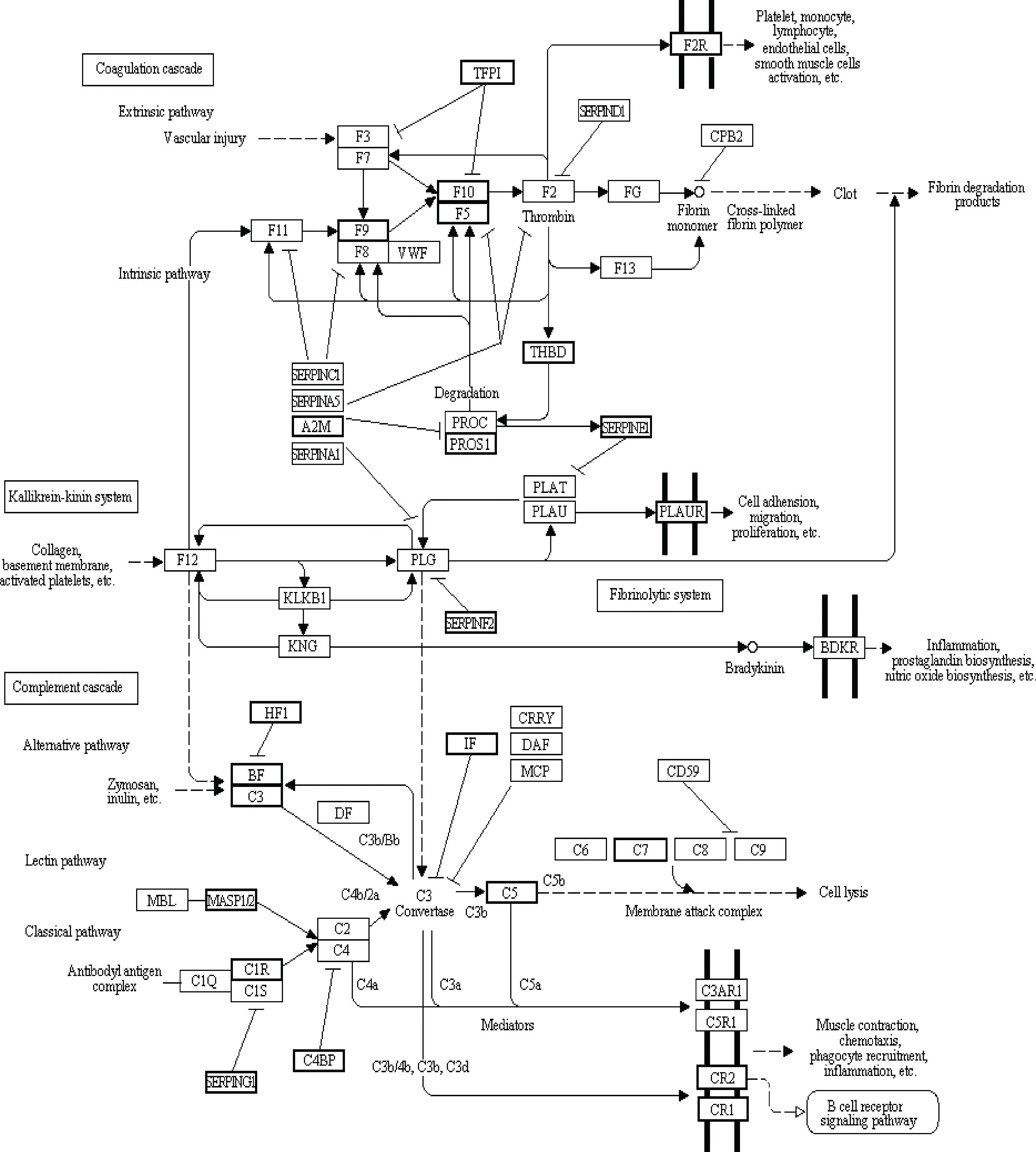
加粗黑框基因为差异表达基因。下同Bold boxes indicate the genes that were differentially expressed.The same as below图1 补体和凝血级联通路Fig.1 Differentially expressed genes in the complement and coagulation cascades pathways
一些与卵巢生殖功能无显著关联的通路也出现在富集水平前10的通路中,比如 “昼夜节律”(Circadian rhythm)通路。不同生物的生理和代谢活动以及行为均遵循着某种昼夜节律,这种特殊的机制称之为“生物钟”。该机制的分子基础有赖于相关生物钟基因的协调表达;产生的生物钟蛋白会形成一个负向转录-翻译反馈环路,这个环路维持着昼夜节律并帮助哺乳动物适应环境的变化[37]。哺乳动物中已确定的主要生物钟蛋白包括CLOCK蛋白、BMALL蛋白、CRY家族以及PER家族,其中CLOCK和BMALL蛋白在昼夜节律调节中处于核心地位[38]。本研究中,“昼夜节律”通路出现富集,且与黄牛数据相比,牦牛的CLOCK基因表达量下调(图2)。笔者推测,这可能与牦牛生活的特殊环境有关。由于青藏高原的高海拔、缺氧环境与强紫外线,牦牛对环境条件如温度和光线的微弱波动感受更加敏感。同时牦牛的繁育活动具有典型的季节性特征,易受到季节性变化的影响,这可能也与该通路具有相关联系。该通路在牦牛卵巢中的确切功能,仍需进一步研究证实。
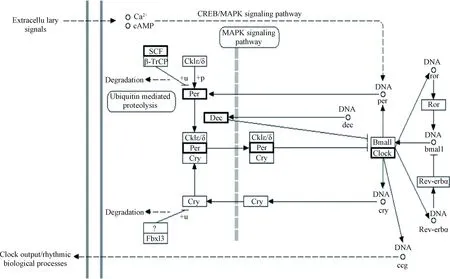
图2 昼夜节律通路Fig.2 Differentially expressed genes in the circadian rhythm pathway
4 结 论
应用 RNA-Seq 技术首次对牦牛和黄牛发情期卵巢进行了转录组学比对分析,共筛选出1 307个显著差异表达基因,同时对这些差异表达基因进行相关功能分析。本研究结果为进一步了解牦牛卵巢的基本分子机制,以及牦牛繁殖的特殊性提供分子基础。
[1]QIU Q,ZHANG G,MA T,et al.The yak genome and adaptation to life at high altitude[J].NatGenet,2012,44(8):946-949.
[2]XIONG X,FU M,LAN D,et al.Yak response to high-altitude hypoxic stress by altering mRNA expression and DNA methylation of hypoxia-inducible factors[J].AnimBiotechnol,2015,26(3):222-229.
[3]MIAO F,GUO Z,XUE R,et al.Effects of grazing and precipitation on herbage biomass,herbage nutritive value,and yak performance in an alpine meadow on the Qinghai-Tibetan Plateau[J].PLoSOne,2015,10(6):e0127275.
[4]FU M,CHEN Y,XIONG X,et al.Establishment of mammary gland modelinvitro:culture and evaluation of a yak mammary epithelial cell line[J].PLoSOne,2014,9(12):e113669.
[5]GUO X,LONG R,KREUZER M,et al.Importance of functional ingredients in yak milk-derived food on health of Tibetan nomads living under high-altitude stress:a review[J].CritRevFoodSciNutr,2014,54(3):292-302.
[6]LONG R J,ZHANG D G,WANG X,et al.Effect of strategic feed supplementation on productive and reproductive performance in yak cows[J].PrevVetMed,1999,38 (3):195-206.
[7]PRAKASH B S,SARKAR M,MONDAL M.An update on reproduction in yak and mithun[J].ReprodDomestAnim,2008,43(Suppl 2):217-223.
[8]FU M,XIONG X R,LAN D L,et al.Molecular characterization and tissue distribution of estrogen receptor genes in domestic yak[J].Asian-AustralasJAnimSci,2014,27(12):1684-1690.
[9]XIAO X,ZI X D,NIU H R,et al.Effect of addition of FSH,LH and proteasome inhibitor MG132 toinvitromaturation medium on the developmental competence of yak (Bosgrunniens) oocytes[J].ReprodBiolEndocrinol,2014,12:30.
[10]PAN L,GONG W,ZHOU Y,et al.A comprehensive transcriptomic analysis of infant and adult mouse ovary[J].GenomicsProteomicsBioinformatics,2014,12(5):239-248.
[11]MIAO X,LUO Q.Genome-wide transcriptome analysis between small-tail Han sheep and the Surabaya fur sheep using high-throughput RNA sequencing[J].Reproduction,2013,145(6):587-596.
[12]CUI Y,YU S J.An anatomical study of the internal genital organs of the yak at different ages[J].VetJ,1999,157(2):192-196.
[13]BECKER J,HACKL M,RUPP O,et al.Unraveling the Chinese hamster ovary cell line transcriptome by next-generation sequencing[J].JBiotechnol,2011,156(3):227-235.
[14]权凯,张兆旺.母牦牛的繁殖特性[J].畜牧与饲料科学,2005,26(5):30-32.
QUAN K,ZHANG Z W.Reproductive characteristics of female yak[J].AnimalHusbandryandFoodScience,2005,26(5):30-32.(in Chinese)
[15]ZI X D,HE S M,LU H,et al.Induction of estrus in suckled female yaks (Bosgrunniens) and synchronization of ovulation in the non-sucklers for timed artificial insemination using progesterone treatments and Co-Synch regimens[J].AnimReprodSci,2006,92(2):183-192.
[16]HU Q,MA T,WANG K,et al.The yak genome database:an integrative database for studying yak biology and high-altitude adaption[J].BMCGenomics,2012,13:600.
[17]AUDIC S,CLAVERIE J M.The significance of digital gene expression profiles[J].GenomeRes,1997,7(10):986-995.
[18]EPPIG J J.Intercommunication between mammalian oocytes and companion somatic cells[J].Bioessays,1991,13(11):569-574.
[19]GILCHRIST R B,RITTER L J,ARMSTRONG D T.Oocyte-somatic cell interactions during follicle development in mammals[J].AnimReprodSci,2004,83:431-446.
[20]MAKKI M,SABOORI E,SABBAGHI M A,et al.Effects of selenium,calcium and calcium ionophore on human oocytesinvitromaturation in a chemically defined medium[J].IranJReprodMed,2012,10(4):343-348.
[21]ROZINEK J,RAJMON R,PETR J,et al.Ultrastructural localisation of calcium deposits in pig ovarian follicles[J].AnimReprodSci,2006,91(2):123-132.
[22]HEDIGER M A.Membrane permeability.The diversity of transmembrane transport processes[J].CurrOpinCellBiol,1997,9(4):543-546.
[23]KIM A M,BERNHARDT M L,KONG B Y,et al.Zinc sparks are triggered by fertilization and facilitate cell cycle resumption in mammalian eggs[J].ACSChemBiol,2011,6(7):716-723.
[24]ANCHORDOQUY J M,ANCHORDOQUY J P,SIRINI M A,et al.The importance of having zinc duringinvitromaturation of cattle cumulus-oocyte complex:role of cumulus cells[J].ReprodDomestAnim,2014,49(5):865-874.
[25]AZUMA T,KONDO T,IKEDA S,et al.Effects of EDTA saturated with Ca2+(Ca-EDTA) on pig,bovine and mouse oocytes at the germinal vesicle stage during maturation culture and the involvement of chelation of Zn2+in pronuclear formation induction by Ca-EDTA[J].Reproduction,2002,124(2):235-240.
[26]SUNYER J O,BOSHRA H,LORENZO G,et al.Evolution of complement as an effector system in innate and adaptive immunity[J].ImmunolRes,2003,27(3):549-564.
[27]APPLEDORN D M,MCBRIDE A,SEREGIN S,et al.Complex interactions with several arms of the complement system dictate innate and humoral immunity to adenoviral vectors[J].GeneTher,2008,15(24):1606-1617.
[28]MOISEYEV G,GIVLI S,BAR-YOSEPH P Z.Fibrin polymerization in blood coagulation-a statistical model[J].JBiomech,2013,46(1):26-30.
[29]HARRIS C L,MIZUNO M,MORGAN B P.Complement and complement regulators in the male reproductive system[J].MolImmunol,2006,43(2):57-67.
[30]MASTELLOS D,GERMENIS A E,LAMBRIS J D.Complement:an inflammatory pathway fulfilling multiple roles at the interface of innate immunity and development[J].CurrDrugTargetsInflammAllergy,2005,4(1):125-127.
[31]HASTY L A,LAMBRIS J D,LESSEY B A,et al.Hormonal regulation of complement components and receptors throughout the menstrual cycle[J].AmJObstetGynecol,1994,170(1):168-175.
[32]DANIELSON P B.The cytochrome P450 superfamily:biochemistry,evolution and drug metabolism in humans[J].CurrDrugMetab,2002,3(6):561-597.
[33]KANDIEL M M,WATANABE G,TAYA K.Ovarian expression of inhibin-subunits,3beta-hydroxysteroid dehydrogenase,and cytochrome P450 aromatase during the estrous cycle and pregnancy of Shiba goats (Caprahircus)[J].ExpAnim,2010,59(5):605-614.
[34]SAHMI M,NICOLA E S,PRICE C A.Hormonal regulation of cytochrome P450 aromatase mRNA stability in non-luteinizing bovine granulosa cellsinvitro[J].JEndocrinol,2006,190(1):107-115.
[35]FRADETTE C,DU SOUICH P.Hypoxia-inducible factor-1 and activator protein-1 modulate the upregulation of CYP3A6 induced by hypoxia[J].BrJPharmacol,2003,140(6):1146-1154.
[36]SIMONSON T S,YANG Y,HUFF C D,et al.Genetic evidence for high-altitude adaptation in Tibet[J].Science,2010,329(5987):72-75.
[37]EDERY I.Circadian rhythms in a nutshell[J].PhysiolGenomics,2000,3(2):59-74.
[38]PIGGINS H D.Human clock genes[J].AnnMed,2002,34(5):394-400.
(编辑程金华)
Comparative Transcriptome Analysis between Yak and Cattle Estrus Ovary
LAN Dao-liang1,2,XIONG Xian-rong2,CHAI Zhi-xin1,AI Yi1,HUANG Cai2,LI Jian1,2*
(1.InstituteofQinghai-TibetanPlateau,SouthwestUniversityforNationalities,Chengdu610041,China;2.CollegeofLifeScienceandTechnology,SouthwestUniversityforNationalities,Chengdu610041,China)
For elaborate the general physiological molecular features of the estrus ovary of yaks and particularity of yak reproduction.RNA-seq technology was applied to analyze transcriptome data comparatively between the yak and plain cattle estrus ovaries in this study.Compared with cattle,genome-wide divergent expression patterns during the ovary transcriptome data revealed that 1 307 genes were significantly and differentially expressed,in which 661 genes were upregulated and 646 genes were downregulated.Functional analysis showed that the differentially expressed genes were involved in various GO categories and KEGG pathways.GO annotations indicated that the genes were closely related to cell adhesion,hormonal biological processes,whereas the calcium ion binding,cation transmembrane transport molecular events were significantly active.KEGG pathway analysis showed that the complement and coagulation cascade pathway was the most enriched,followed by the cytochrome P450 related pathways.Moreover,several novel pathways,such as circadian rhythm,were significantly enriched despite having no evident associations with the reproductive function.This study is the first to compare the ovary transcriptome data between yak and cattle and to identify and analyze the related differential genes.Our findings provide a theory support for further investigation of the molecular mechanism of yak ovary and offer new insights to understand comprehensively the specificity of yak reproduction.
yak;estrus ovary;transcriptome;comparative analysis;general molecular mechanism
10.11843/j.issn.0366-6964.2016.09.011
2016-03-31
西南民族大学中央高校基本科研业务费专项资金资助(2015NZYTD01)
兰道亮(1984-),男,畲族,江西德兴人,副研究员,博士,主要从事高原动物遗传育种研究,E-mail:landaoliang@163.com;Tel:028-85522552
李键,教授,E-mail:lijian@swun.cn
S823.8+5.2
A
0366-6964(2016)09-1830-10
