一个临床表现迥异的伴皮质下囊肿的巨脑性白质脑病家系
2016-10-31魏妍平崔丽英
魏妍平, 崔丽英, 彭 斌
一个临床表现迥异的伴皮质下囊肿的巨脑性白质脑病家系
魏妍平,崔丽英,彭斌
目的报道一个伴皮质下囊肿的巨脑性白质脑病(MLC)家系兄妹两人,并结合文献复习,以期对MLC的早期诊断提供帮助。方法分析这2例患者的临床表现,影像学资料,基因检查以及其它辅助检查结果。结果本文2例患者为同一家系兄妹两人,临床症状差异显著,但影像学均表现典型的白质病变和皮质下囊肿,MLC基因均发现两处杂合突变:c. 812T>C,c. 250C>G。结论MLC患者影像学表现具有相对特异性,而临床症状变化多样,详细询问家族史,并进行相关基因检测,才能尽早明确诊断。
皮质下囊肿的巨脑性白质脑病;vanderKnaap病;核磁共振
1995年荷兰医生VanderKnaap首先报道伴皮质下囊肿的巨脑性白质脑病(MegalencephalicLeukoencephalopathywithsubcorticalCystsMLC),因此该病又称为vanderKnaap病,属于常染色体隐性遗传病[1]。患者一般于儿童期起病,中位发病年龄为6 m,多于1岁之内出现巨颅,此后主要表现为运动发育落后和癫痫发作,可出现锥体束、锥体外系和小脑受累的表现,智力障碍相对较轻[2,3]。MLC的诊断主要基于临床症状和影像学,典型的头部核磁共振(MRI)呈现弥漫性脑白质病变,伴有颞极和(或)前额部皮质下囊肿[2,3]。本文报道MLC一家系兄妹两人,临床症状迥异,但两者头部MRI均符合典型的MLC影像学表现,MLC基因二代序均发现复合杂合突变。
1 临床资料
先证者,女,22岁,因间断头痛,左侧肢体感觉异常1 m来诊。患者生长发育如常,1岁半时能独立行走。近1 m间断头痛,较剧烈,左侧肢体紧箍感,肌力未下降。既往20岁时于外伤后出现步态异常,日常生活不受限,近期未加重。初中一年级退学,学习成绩差。查体:头围62 cm,言语流利,脑神经检查基本正常。计算力、记忆力、理解判断力减退。四肢肌力5级,肌张力高,双侧高足弓,足内翻,踝关节挛缩固定。双下肢病理征阳性,痉挛性步态。感觉检查基本正常。头部MRI可见双侧半卵圆中心、侧脑室旁长T1、长T2异常信号,双侧额颞叶皮质下可见多发囊肿(见图1),基底节和丘脑无明显异常。血氨基酸和酰基肉碱谱、尿有机酸、血芳基硫酸酯酶A、血半乳糖脑苷脂酶测定均正常。血乳酸、肝功、肾功正常。心电图、超声心动图正常。MLC基因外显子区域发现两处杂合突变点:c. 812T>C,c. 250C>G,导致氨基酸改变,p. L271P,p. R84G(亮氨酸>脯氨酸,精氨酸>甘氨酸)。Sanger测序验证chr22-50506944存在c. 812T>C的杂合突变,chr22-50521530存在c. 250C>G的杂合突变。检索HGMDpro数据库,未见报道。半年后随访,患者病情无变化。
先证者之兄(未就诊),自幼头围大,具体不详。运动发育落后,行走困难。7岁后智力、语言较同龄儿差,并且反复癫痫发作。现在28岁,不能独立行走,只能说单字。头部MRI与先证者类似,可见白质弥漫性病变和额颞叶皮质下囊肿形成(见图2)。Sanger测序存在c. 812T>C的杂合突变和c. 250C>G的杂合突变,与先证者相同。先证者父母及其他亲属未发现类似症状。先证者之母c. 250C>G杂合突变。其父45岁时去世,无类似症状。
2 讨 论
本文2例患者为同一家系兄妹两人,临床症状迥异。先证者症状轻微,除行走较同龄儿稍晚外,其他生长发育基本正常;成年后,隐袭起病,步态障碍,无明显加重,日常生活完全自理。近1 m出现间断头痛、左侧肢体感觉异常。查体发现认知功能减退,双侧锥体束受损,无明确感觉障碍。先证者临床表现无特异性,可见于多种遗传代谢病。其兄症状严重,自幼头围大,生长发育异常,智力和运动发育迟滞并倒退,反复癫痫发作,符合文献中MLC经典型的临床表现。2例患者头部MRI均为弥漫性白质病变,同时可见双侧顶叶、颞叶皮质下囊肿形成,符合MLC的典型影像学表现。此外,先证者血氨基酸和酰基肉碱谱、尿有机酸、血芳基硫酸酯酶A、血半乳糖脑苷脂酶测定均正常,可排除有机酸尿症、异常氨基酸血症、异染性脑白质营养不良、球样细胞脑白质营养不良等。血乳酸正常不支持线粒体病。其他需要鉴别的白质脑病,如Canavan病、亚历山大病、merosin缺乏的先天性肌营养不良、GM1或GM2神经节苷脂贮积症等,均与先证者的临床症状与影像学不符,可逐一排除。据此,本文2例患者符合MLC的临床诊断。
MLC的诊断主要根据临床表现和头部MRI所见,但MRI表现与临床症状可以不平行,甚至分离[3]。MLC主要的临床表现为头围增大,尤其是1岁之内最明显;运动发育迟缓或倒退,甚至丧失行走能力,可出现手足徐动或肌张力障碍等锥体外系受累的症状;构音障碍进行性加重,甚至完全失语;智力障碍常在病程后期出现,相对较轻;多数患者出现癫痫发作,但对药物反应敏感;轻微脑外伤常导致患者病情急性加重[2,3]。本文同一家系兄妹两人,MLC的临床表现差异显著,兄长发病早,症状典型,容易诊断,而先证者临床症状轻,容易误诊。此时详细询问家族史和生长发育史,非常重要。MLC头部MRI主要表现为弥漫的大脑白质病变,同时伴有双侧颞叶、额叶前部、额顶交界区,特征性的皮质下囊肿,具有重要的诊断价值[2~4]。MLC早期白质病变以水肿为主,囊肿很小或尚未形成,后期出现白质萎缩,囊肿体积增大,数目增多[3,4]。本文2例患者头部MRI完全符合MLC患者的典型表现,是提示进一步基因检查的线索。
2001年Leegerwater等首先提出MLC的致病基因位于22q13.3,并命名为MLC1基因[5]。MLC1基因包含12个外显子,编码含377个氨基酸的MLC1蛋白。2011年Lopez-Hernandez等发现了第二个MLC的致病基因,即胶质细胞粘附分子基因(glial cell adhesion molecule,GlialCAM)[6]。约75%的患者为MLC1基因突变,约20%为GlialCAM突变,另约5%患者致病基因不明[4]。MLC1基因纯合或复合杂合突变所导致的MLC又称为MLC1,临床表现为经典型,为常染色体隐性遗传。而GlialCAM相关的MLC又称为MLC2,并分为MLC2A和MLC2B。前者为常染体隐性遗传,临床表现为经典型,而后者为常染色体显性遗传,临床表现为改善型,即有些患者的临床症状可能改善,甚至消失,头部MRI可能恢复正常[6]。MLC1蛋白和GlialCAM都属于跨膜蛋白,共同存在于星形细胞的终足,可能与离子通道、转运有关,在脊椎动物中属于高度保守的蛋白[6]。
2例患者MLC1基因二代测序,Sanger验证均发现相同的复合杂合突变,c. 812T>C和c. 250C>G,并且证实c. 250C>G位点突变来自于无症状的母亲。遗憾的是其父亲早逝,未能采集其血样。但在千人基因组,以及in house数据库中,携带率为零。计算机软件预测突变的致病性结果发现:SIFT软件预测为有害,PolyPhen-2预测为很可能有害,Mutation Taster预测为致病突变。综上所述,此复合杂合突变很可能是致病突变。
MLC发病率低,既往文献也多为零星病例报道,本文2例患者的特点在于:(1)先证者症状轻微,容易误诊,提示对于不能解释而且起病隐袭的神经系统症状,即使成年发病,也应详细询问家族史和生长发育史,避免误诊。(2)2例患者虽然症状不同,但影像学表现相似,提示遗传性疾病可能,并且根据相对特异的MRI表现,进行相关基因检测,最终明确诊断。(3)发现新的复合杂合突变,很可能是致病突变。
总之,对疑似MLC患者进行MLC1基因分析,不但可以明确大部分患者的致病突变,从而进行遗传咨询和产前诊断,而且有助于进一步研究MLC的发病机制,为基因治疗提供佐证。
[1]Van der Knaap MS,Barth P,Stroink H,et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children[J]. Ann Neurol,1995,37:324-334.
[2]Singhal BS,Gorospe JR,Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts[J]. Journal of Child Neurology,2003,18:646-652.
[3]Patrono C,Di Giacinto G,Eymarde Pierre E,et al. Genetic heterogeneity of megalencephalic leukoencephalopathy and subcortical cysts[J]. Neurology,2003,61:534-537.
[4]Van der Knaap MS,Boor I,Estevez R. Megalencephalic leukoencephalopathy with subcortical cysts:chronic white matter oedema due to a defect in brain ion and water homoeostasis[J]. Lancet Neurology,2012,11:973-985.
[5]Leegwater P,Yuan B,van der Steen J,et al. Mutations of MLC1(KIAA0027),encoding a Hum putative membrane protein,cause megalencephalic leukoencephalopathy with subcortical cysts[J]. Am J Genet. ,2001,68:831-838.
[6]Lopez-Hernandez T,Ridder MC,Montolio M,et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts,benign familial macrocephaly,and macrocephaly with retardation and autism[J]. American Journal of Human Genetics,2011,88:422-432.
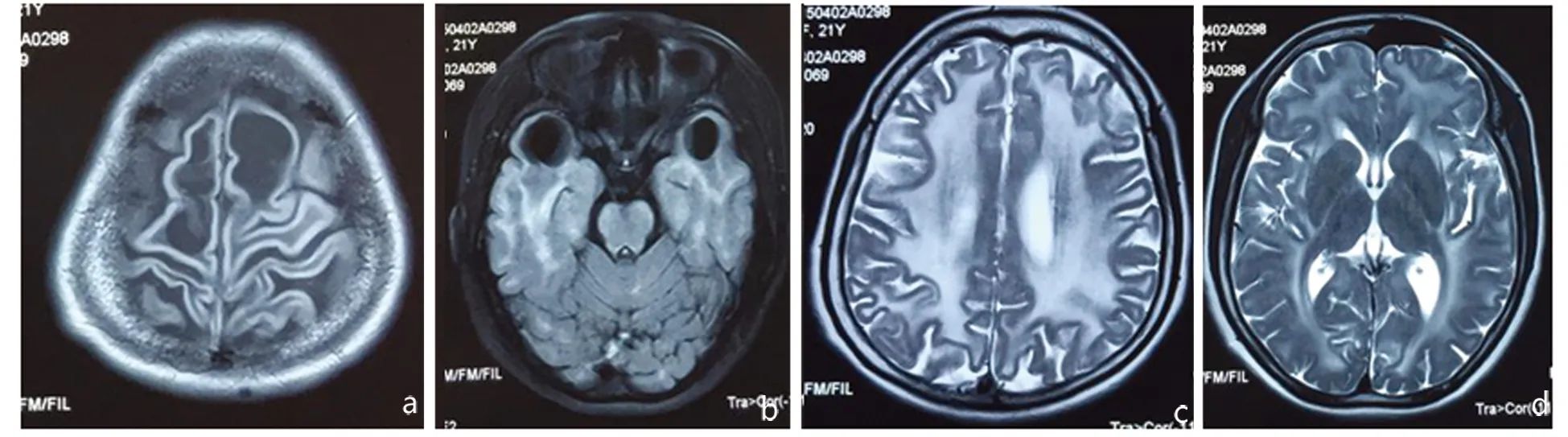
图1头部MRI可见双侧顶叶和双侧颞叶皮质下囊肿(1a,1b,Flair相);双侧半卵圆中心、脑室旁白质以及皮质下白质弥漫性长T2异常信号(1c,1d,T2相)
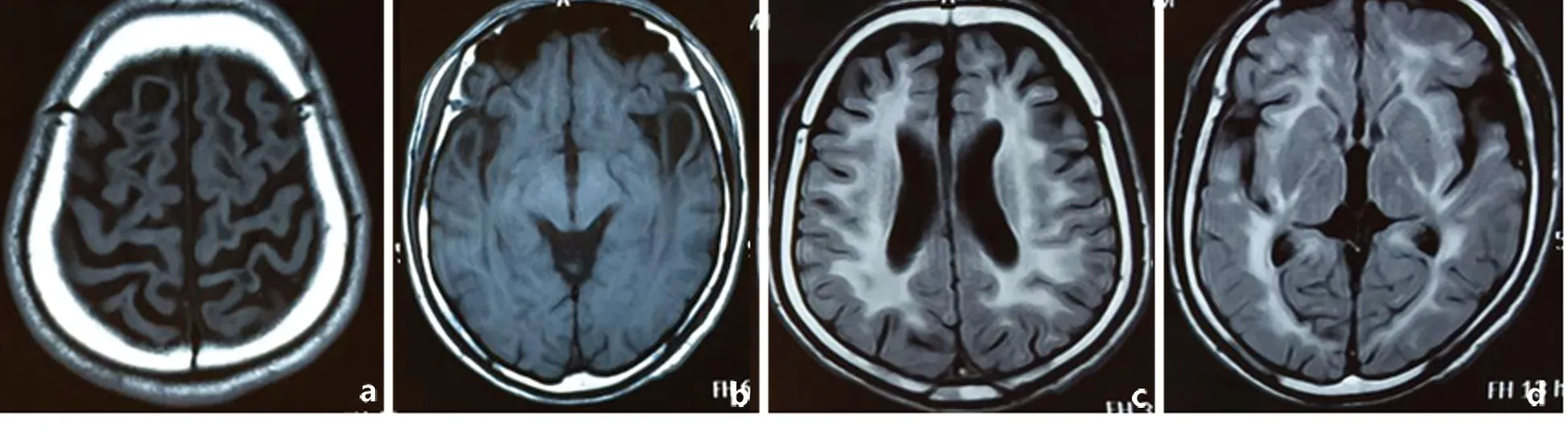
图2头部MRI可见双侧顶叶和双侧颞叶皮质下囊肿(2a,2b,T1相,);双侧脑室旁白质以及皮质下白质弥漫性异常信号(2c,2d,Flair相)
A pedigree of megalencephalic leukoencephalopathy with cysts presenting with distinct clinical features
WEIYanping,CUILiying,PENGBin.
(DepartmentofNeurology,PekingUnionMedicalCollegeHospital,ChineseAcademyofMedicalScience,Beijing100730,China)
ObjectiveMegalencephalic leukoencephalopathy with cysts (MLC) is a kind of autosomal recessively inherited disease which is relatively rare,but with distinct clinical symptoms and neuroimage. A pedigree of MLC was reported,meanwhile,related documents were reviewed,in order that early diagnosis was made easier. MethodsA brother and a sister from the MLC pedigree were analyzed including clinical features,neuroimage,the other ancillary examinations and genetic test. ResultTwo MLC cases were from the same pedigree with definitely different clinical presentation while their MRI image indicated the same white matter lesion with subcortical cyst formation. Two heterogenous point mutations,c. 812T>C,c. 250C>G,were found from MLC1 gene. ConclusionThe clinical presentations of MLC may be variable while the neuroimage generally are characteristic which could be one of the diagnostic clues. Early diagnosis of MLC was only achieved with thorough inquiry of family history and related gene test.
Megalencephalic leukoencephalopathy with cysts;Van der Knaap disease;Magnetic resonance imaging
1003-2754(2016)09-0776-03
2016-07-20;
2016-08-03
(中国医学科学院北京协和医学院北京协和医院神经内科,北京 100730)
魏妍平,E-mail:yp924@sina.com
R742
A
