Si-B-C-N-H-Cl体系CVD气相产物热力学数据的计算
2016-10-12任海涛刘家臣郭安然天津大学先进陶瓷加工技术教育部重点实验室天津30007西北工业大学超高温结构复合材料重点实验室西安7007
任海涛,刘家臣,郭安然天津大学先进陶瓷加工技术教育部重点实验室,天津30007西北工业大学超高温结构复合材料重点实验室,西安7007
Si-B-C-N-H-Cl体系CVD气相产物热力学数据的计算
任海涛1,2,刘家臣1,郭安然11天津大学先进陶瓷加工技术教育部重点实验室,天津300072
2西北工业大学超高温结构复合材料重点实验室,西安710072
本文采用基于量子力学的理论方法计算获得了Si-B-C-N-H-Cl 体系CVD过程中所有气相产物的热力学数据,包括分子在298.15 K ~ 2000 K的标准摩尔热容,标准摩尔熵、标准摩尔生成焓和标准摩尔生成吉布斯自由能等,为含Si-B-C-N-H-Cl元素的任意先驱体体系反应热力学研究提供了基础数据。
硅硼碳氮陶瓷;CVD;热力学计算
连续纤维增韧非氧化物陶瓷基复合材料具有密度低、耐高温、优异力学、抗腐蚀和抗氧化性能,在航空航天领域获得了广泛应用。含有硅 (Si)、硼 (B)、碳 (C)、氮 (N) 等化学元素的二元或多元非氧化物陶瓷 (如 SiC、Si3N4、BN、Si-C-N和 Si-B-C-N 等) 具有轻质、耐高温、抗热冲击、力学性能高等优点,其中一些化合物还具有吸波透波功能,是结构功能一体化陶瓷基复合材料的优良基体材料[1-3]。
化学气相沉积 (Chemical Vapor Deposition, CVD) 是制备陶瓷基复合材料界面、基体和涂层组元的有效方法。利用CVD方法制备无机材料的反应过程中涉及到的化学反应十分复杂,理解和掌握这些反应的机理对于先驱体的选择、反应器的设计、工艺参数的优化以及材料的微观尺度设计都有极其重要的意义。Si-B-C-N-H-Cl体系中涉及到大量的气相产物,其中绝大多数气相产物由于存在寿命短,活性高,实验难以测定,其热力学数据基本都为未知,仅通过传统实验方法研究其热力学特性目前还很困难。
西北工业大学苏克和教授课题组基于量子化学理论结合精确模型化学方法分别对丙烯热解制备热解碳体系[4,5]、MTS热解制备碳化硅 (SiC) 体系[6-8]、BCl3-CH4-H2-Ar制备碳化硼(B4C)体系[9,10]以及BCl3-C3H6-H2-Ar制备碳化硼 (B4C) 体系[11,12]进行了研究。基于体系Gibbs自由能最小的方法是最常用的热力学计算方法,而其研究基础是可靠的高温热力学数据。
本文利用量子化学结合统计热力学方法,计算获得Si-B-C-N-H-Cl体系中相关气相产物的热力学数据,其中包括Si-B体系[13]的211种、B-N体系[14]的136种、Si-N体系[15]的155种、Si-C-N体系[16]的438种。这些数据包括各气相产物的标准摩尔热容Cθp、标准摩尔熵Sθm、生成焓ΔfHmӨ和生成吉布斯自由能ΔfGmӨ。可为含Si、B、C、N、H、Cl元素的任意先驱体体系CVD过程反应热力学研究提供基础数据。选择 JANAF[17]中可查到实验数据的 16 种气相产物为例说明。
1 气相产物热力学计算方法
本文采用标准统计热力学方法来计算不同温度下的热容和熵,考虑了平动配分函数、转动配分函数、振动配分函数及电子配分函数对热容和熵的贡献。具体计算细节如下:
1.1配分函数
平动配分函数:当分子平动能级差很小时,平动配分函数qt的表达式为:
式中,m为分子的质量,V为分子的体积,T为温度,k为玻尔兹曼常数,h为普朗克常数。
根据阿伏伽德罗公式pV = nRT = NAkT (其中p为气体压强,R为气体常数,NA为阿伏伽德罗常数),可知V = NAkT/p,代入式 (1) 即得到:

转动配分函数:同样,当转动能级差很小时,分子的转动配分函数qr有解析式。对于直线型分子,其解析式为:
式中,I为转动惯量,σ为对称数。
振动配分函数:因分子的振动能级差较大,振动激发遵循统计规律。分子的振动配分函数qν也分直线型和非直线型分子。取分子的振动基态 (零点振动能级) 为能量零点,则直线型分子的配分函数为:

对于非直线型分子则为:

式中,n为分子所含原子数,(3n - 5) 或 (3n - 6) 为振动自由度或独立振动模式数 (因为n原子分子在三维空间运动的总自由度为3n,其中分子质心平动自由度为3,直线分子转动为2,非直线分子转动为3,故3n - 5或3n - 6即为振动自由度数),vi为分子中第i种振动模式的振动频率 (Hz)。
电子配分函数:电子激发能随分子的不同而不同。在CVD/CVI方法制备材料所通常采用的较高温度下,电子激发是一个不容忽视的问题,必须在热力学数据计算中加以考虑。电子配分函数qe的表达式如下:

式中,gi是第i个电子能态的量子权重 (或简并度),εi是第i个态的能量。简并度按以下方法计算:对于单原子,简并度gi= 2J + 1,J为电子总 (旋轨耦合) 轨道角动量量子数;对于多原子分子,简并度即为自旋多重度与各激发态所属点群的不可约表示维数之积。对于个别分子,由于一些算法的缺陷,会得到一个与基态能量很接近的准激发态。本文取其和基态的简并度各占基态总简并度的一半。
1.2热熔和熵
考虑到平动、转动、振动及电子配分函数对热容和熵的贡献,气态单原子的总热容 (CӨp,m) 和熵) 的表达式为:
式中,B = h/8cπ2I,c是真空中的光速,μ =ω/(kT),ω是谐振子的基频。气态线性多原子分子的总热容 (CӨp,m) 和熵 (SӨm) 的表达式为:
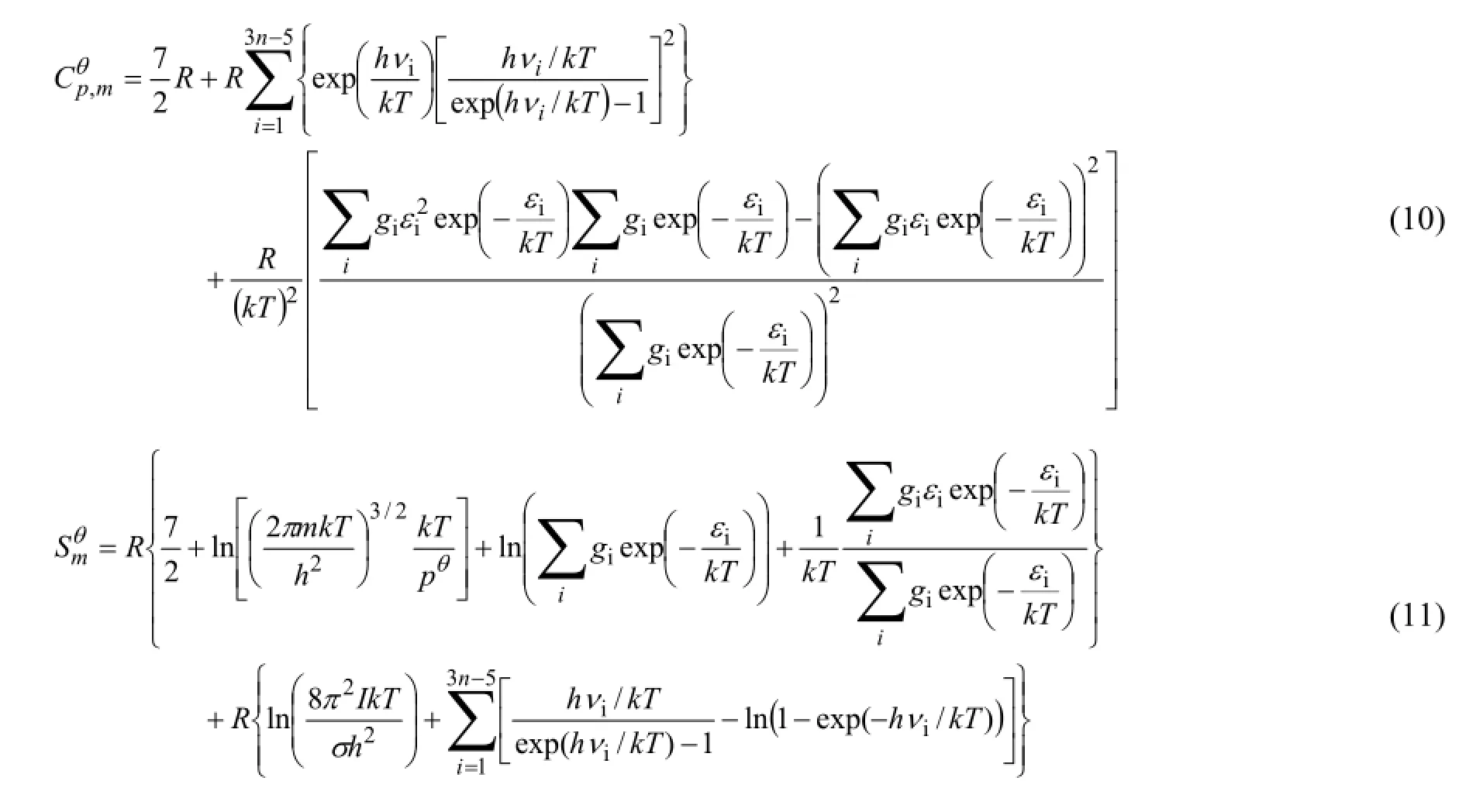
气态非线性多原子分子的总热容 (CӨp,m) 和 熵(SӨm) 的表达式为:
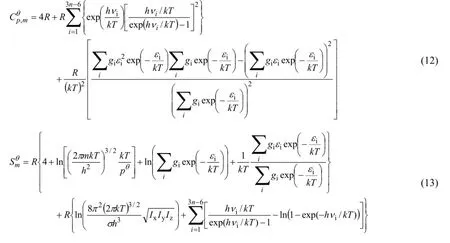
1.3标准生成焓和标准生成吉布斯自由能
标准生成焓 ΔfHmθ和标准生成吉布斯自由能 ΔfGmθ通过原子化反应 [式 (14)] 计算。具体计算公式见式 (15) 及式 (16):

其中,μi代表第i个物种的化学计量数,原子i的ΔfHmӨ(i,g,T) 和ΔfGmӨ(i,g,T) 是从JANAF[17](或CODATA[18]) 中查到的实验数据。式中的ΔrHmӨ(T) 和ΔrGmӨ(T) 是由以下公式计算得到的反应焓变及反应吉布斯自由能变:
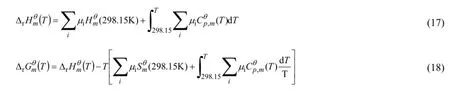
其中,HmӨ(298.15K) 是由G3(MP2) 和G3//B3LYP方法计算的电子能量结合统计热力学处理所得298.15K下的标准焓,CӨp,m(T) 是标准摩尔热容拟合结果,SmӨ(298.15K) 是298.15K下的标准熵。
2 结果与讨论
2.1分子结构
采用密度泛函 (Density Functional Theory, DFT) 理论,通过Gaussian 09[19]程序,使用B3PW91/6-31G(d) 方法对气相产物 (包括同分异构体) 进行计算,频率如果无负值可认为分子为稳定结构。它们的化学式,分子结构,电子态以及对称性如图1所示。

图1 用B3PW91/6-31G(d) 方法计算得到JANAF中可查到实验数据的 16 种气相产物的化学式、稳定结构、电子态和对称性Figure 1 Numbering, stable structure (symmetry and electronic state) of the 16 species in the JANAF obtained based on B3PW91/6-31G(d) calculations

表1 用B3PW91/6-31G(d)方法计算得到的分子振动频率 (cm-1) 乘以校正因子[20]0.9573和红外光谱强度(km/mol)Table 1 Vibrational frequencies (cm-1) and IR intensities (km/mole) calculated with B3PW91/6-31G(d). The listed frequencies have been scaled by the factor of 0.9573
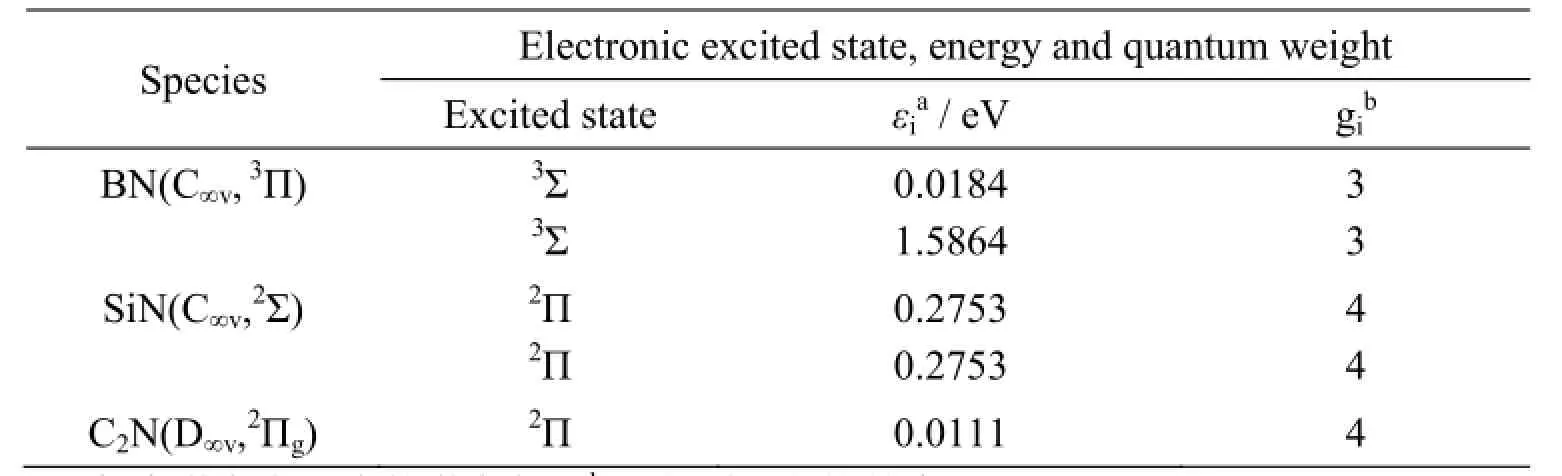
表2 用TD-DFT B3PW91/6-31G(d) 方法计算得到的分子 <1.860 eV的垂直激发能Table 2 Low-lying (<1.860 eV) vertical electronic excitation energies calculated with TD-DFT at B3PW91/6-31G(d) level
2.2振动频率
同样采用B3PW91/6-31G(d) 方法计算得到体系相关气相产物的振动频率 (频率的校正因子[20]为0.9573) 和红外光谱强度,结果如表1所示。可以看到所有分子频率均为正值,说明所有分子都是稳定结构。这些频率将用来计算分子的振动配分函数。
2.3电子激发能
文献 [21] 表明当分子的电子激发能小于1.860 eV时,对于统计热力学计算热熔和熵的影响不可忽略。表2为采用TD-DFT B3PW91/6-31G(d) 方法计算得到的所有气相产物在1.860 eV范围内的激发态,垂直激发能以及简并度。这些激发能将用来计算分子的电子配分函数。
2.4热熔和熵
由前面计算所得数据 (分子结构、振动频率及激发能) 再结合统计热力学,根据式 (8) ~ (13) 便可算出分子在298.15 K的标准摩尔热容Cθp,m和熵Sθm。表3为计算得到的数据以及文献中可查到的实验数据。可见,与文献的实验数据相比,热容值偏差最大是N2H4分子,为4.333 J·mol-1·K-1;熵值偏差最大的是NH分子,为9.223 J·mol-1·K-1;其它分子热容和熵的计算值与实验值符合都很好。

表3 由统计热力学计算得到的298.15 K时分子热容Cθp,m和熵SθmTable 3 Heat capacity Cθp,mand entropy Sθm(298.15 K) calculated based on statistical thermodynamics
2.5分子能量
表4为采用G3(MP2) 和G3//B3LYP方法计算所得分子能量 (即0 K时的热力学能,U0K) 以及在298.15 K分子焓的热修正 (Hcorr) 和分子吉布斯自由能的热修正 (Gcorr)。分子在298.15 K的标准焓值为U0和Hcorr之和,标准吉布斯自由能为U0和Gcorr之和。
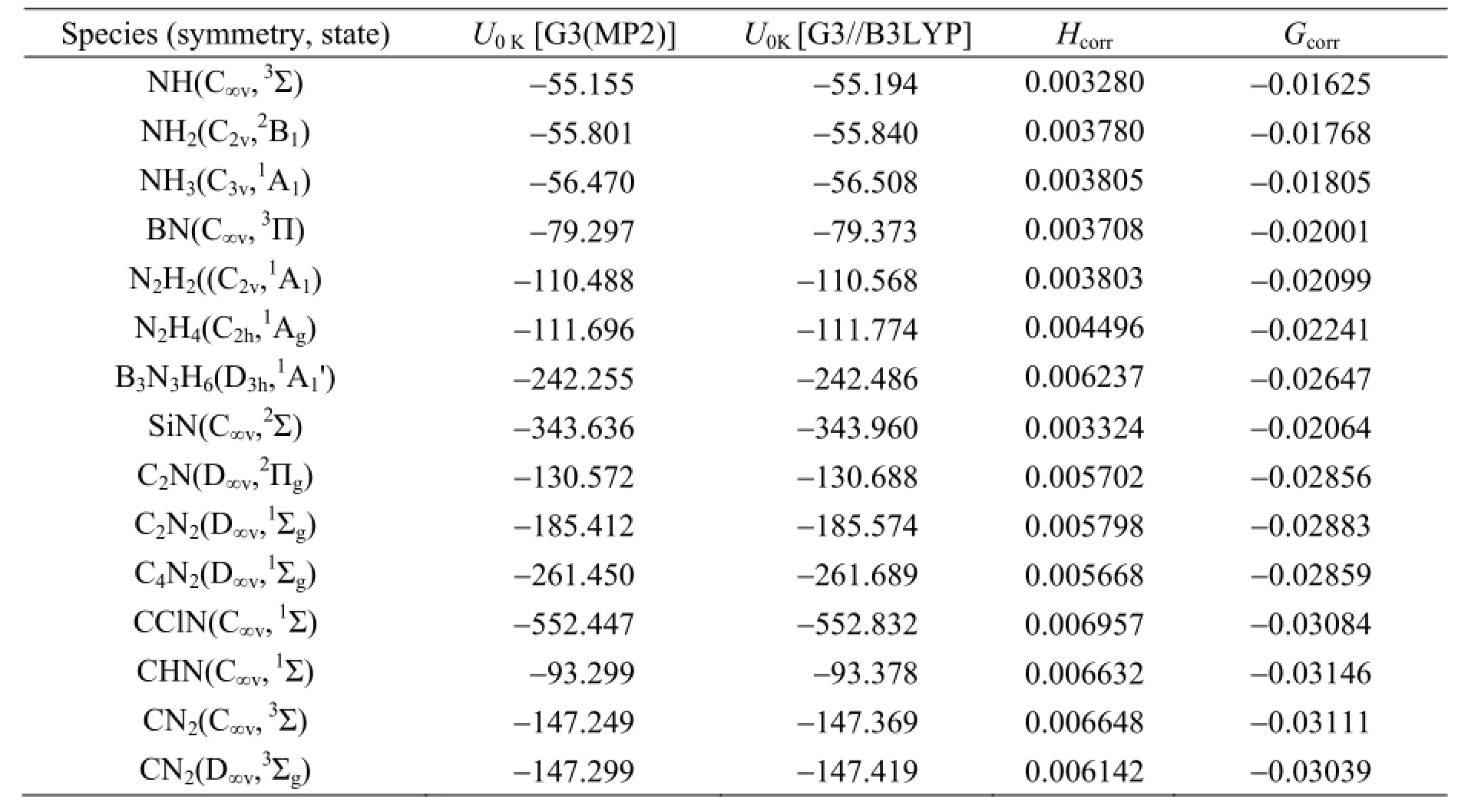
表4 用G3(MP2) 和G3//B3LYP计算得到的分子能量 (U0K) 和热修正 (单位为Hartrees)Table 4G3(MP2) and G3//B3LYP energy (U0K), thermal correction to enthalpy (Hcorr) and to Gibbs free energy(Gcorr) (all in Hartrees) at 298.15K
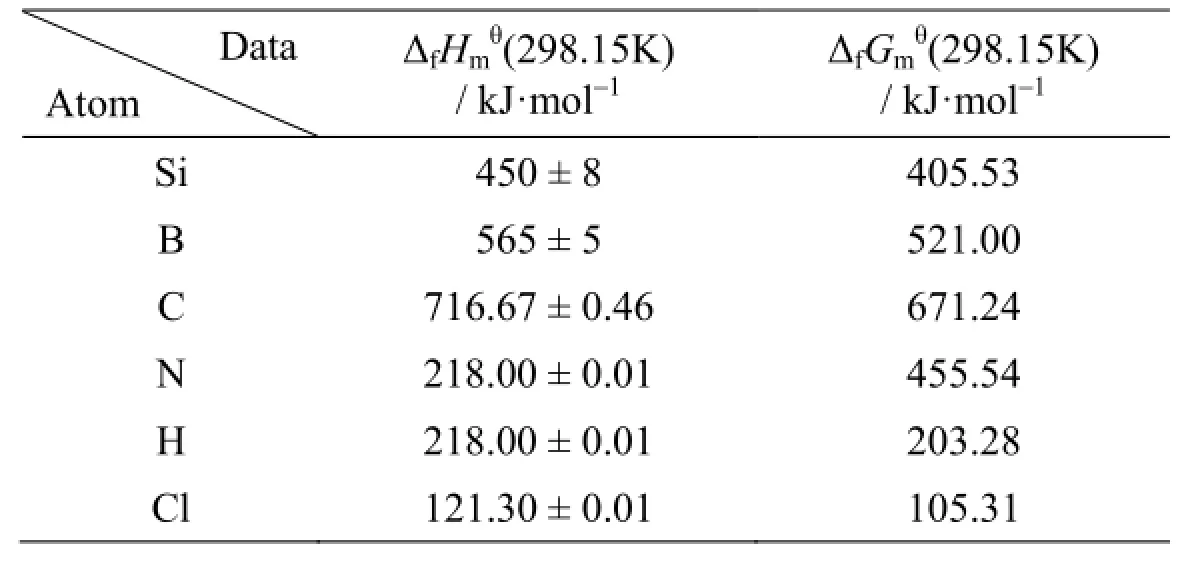
表5 气相Si、B、C、N、H和Cl原子的ΔfHmӨ(298.15 K) 和ΔfGmӨ(298.15 K) 实验数据[17,18]Table 5 The experimental data of ΔfHmӨ(298.15 K) and ΔfGmӨ(298.15 K) of the gaseous Si, B, C, N, H and Cl atoms
2.6标准生成焓和标准生成吉布斯自由能
由式 (15) ~ (18) 可以看出,计算分子的标准生成焓ΔfHmӨ(298.15 K) 和标准生成吉布斯自由能ΔfGmӨ(298.15 K) 必须结合相关原子的实验数据。表5 是所用到的 Si、B、C、N、H 和 Cl 原子的实验数据,其中 B 原子的数据来自 CODATA[18],其它原子的数据均来自 JANAF[17]热力学数据表,这是因为 JANAF给出的B原子生成焓为560 ± 12 kJ·mol-1,不确定度为 ±12 kJ·mol-1,而 CODATA给出的B原子生成焓为565 ± 5 kJ·mol-1,不确定度为 ±5 kJ·mol-1。
根据公式 (15) ~ (18) 结合表3、表4和表5的数据,便可计算出各个分子的标准摩尔生成焓ΔfHmӨ(298.15 K) 和标准摩尔生成吉布斯自由能ΔfGmӨ(298.15 K),分别用G3(MP2) 和G3//B3LYP方法计算所得结果和实验值如表6所示。
由表6可以看出,G3(MP2) 方法和G3//B3LYP方法的计算结果基本一致,除B3N3H6分子相差17.67 kJ·mol-1以外,两种方法的计算差值都在 ±10 kJ·mol-1以内。

表6 G3(MP2) 和G3//B3LYP方法计算得到的分子298.15 K标准摩尔生成焓ΔfHmθ和标准摩尔吉布斯自由能ΔfGmθ与实验值的对比Table 6 Standard enthalpies of formation ΔfHmθ(298.15K) and Gibbs free energies of formation ΔfGmθ(298.15K)predicted with G3(MP2) and G3//B3LYP theories and the comparisons with available experiments
与JANAF数据表中可以查到的实验数据相比,G3//B3LYP方法计算的理论差值分别为24.86,5.172, 2.898, 22.824, 11.334, 17.347,0.53,94.324,117.86,8.682,12.99,7.426,7.44,9.82和27.89 kJ·mol-1。SiN(C∞v,2Σ) 和C2N(D∞v,2Πg) 与实验数据偏差较大,分别为94和117kJ·mol-1。可以看到这两个分子实验数据的误差范围较大,可靠性低。而其它分子的计算结果与实验结果符合都很好。
[1] 张立同, 成来飞, 徐永东. 新型碳化硅陶瓷基复合材料的研究进展 [J]. 航空制造技术, 2003, (1): 24-32.
[2] 张立同, 成来飞. 连续纤维增韧陶瓷基复合材料可持续发展战略探讨[J]. 复合材料学报, 2007, 24 (2): 1-6.
[3] WANG ZC, ALDINGER F, RIEDEL R. Novel silicon-boron-carbon-nitrogen materials thermally stable up to 2200°C [J]. Journal of the American Ceramic Society, 2001, 84 (10): 2179-2183.
[4] 姚小平. 丙烯热解制备PyC的气相反应热力学与Ar@C60包合物力学性能的研究[D]. 西安:西北工业大学硕士学位论文, 2007.
[5] YAO X, SU K, DENG J, et al. Gas-phase reaction thermodynamics in preparation of pyrolytic carbon by propylene pyrolysis [J]. Computational Materials Science, 2007, 40 (4): 504-524.
[6] 邓娟利. CVD/CVI制备自愈合SiC陶瓷基复合材料的反应热力学研究[D]. 西安:西北工业大学博士学位论文, 2009.
[7] DENG J, SU K, ZENG Q, et al. Thermodynamics of the production of condensed phases in the CVD of methyltrichlorosilane pyrolysis [J]. Chemical Vapor Deposition, 2009, 15 (10-12): 281-290.
[8] DENG J, SU K, WANG X, et al. Thermodynamics of the gas-phase reactions in chemical vapor deposition of silicon carbide with methyltrichlorosilane precursor [J]. Theoretical Chemistry Account, 2008, 122(1-2): 1-22.
[9] 曾艳. BCl3-CH4-H2体系CVD 制备碳化硼的气相反应热力学研究[D]. 西安:西北工业大学硕士学位论文, 2008.
[10] ZENG Y, SU K, DENG J, et al. Thermodynamic investigation of the gas-phase reactions in the chemical vapor deposition of boron carbide with BCl3-CH4-H2precursors [J]. Journal of Molecular Structure: THEOCHEM, 2008, 861 (1-3): 103-116.
[11] 王涛. BCl3-C3H6(丙烯)-H2体系CVD 制备碳化硼的气相反应热力学研究[D]. 西安:西北工业大学硕士学位论文, 2008.
[12] WANG T, SU K, DENG J, et al. Reaction thermodynamics in chemical vapor deposition of boron carbides with BCl3-C3H6(propene)-H2precursors [J]. Journal of Theoretical and Computational Chemistry, 2008, 7(6): 1269-1312.
[13] REN H, ZHANG L, SU K, et al. Thermodynamics investigation of the gas-phase reactions in the chemical vapor deposition of silicon borides with BCl3-SiCl4-H2precursors [J]. Structural Chemistry, 2014, 25 (5): 1369-1384.
[14] REN H, ZHANG L, SU K, et al. Thermodynamic study on the chemical vapor deposition of boron nitride from the BCl3-NH3-H2system [J]. Theoretical Chemistry Account, 2014, 133 (11): 1-13.
[15] REN H, ZHANG L, SU K, et al. Thermodynamic study on the chemical vapor deposition of silicon nitride from the SiCl4-NH3-H2system [J]. Computational and Theoretical Chemistry, 2015, 1051: 93-103.
[16] REN H, ZHANG L, SU K, et al. Thermodynamic study of the chemical vapor deposition in the SiCl3CH3-NH3-H2system [J]. Chemical Physics Letters, 2015, 623: 29-36.
[17] CHASE MW. NIST-JANAF thermochemical tables forth edition [J]. Journal of Physical and Chemical Reference Data, 1998, Monograph 9.
[18] HAYNES WM. CRC Handbook of Chemistry and Physics [M]. Boca Raton: CRC Press, 2015.
[19] FRISCH MJ, TRUCKS GW, SCHLEGEL HB, et al. Gaussian 09 [M]. Wallingford, CT, USA, Gaussian,Inc., 2009.
[20] SCOTT AP, RADOM L. Harmonic vibrational frequencies: an evaluation of Hartree-Fock, Møller-Plesset,quadratic configuration interaction, density functional theory, and semiempirical scale factors [J]. The Journal of Physical Chemistry, 1996, 100 (41): 16502-16513.
[21] DENG J, SU K, ZENG Y, et al. Investigation of thermodynamic properties of gaseous SiC (X and a) with accurate model chemistry calculations [J]. Physica A, 2008, 387 (22): 5440-5456.
The Calculation of the Thermodynamic Data of the Gas-Phase in the Chemical Vapor Deposition of the Si-B-C-N-H-Cl System
REN Hai-Tao1,2, LIU Jia-Chen1, GUO An-Ran11Key Laboratory of Advanced Ceramics and Machining Technology of Ministry of Education, School of Materials Science and Engineering, Tianjin University, Tianjin 300072, China2Science and Technology on Thermostructure Composite Materials Laboratory, School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, China
The thermodynamic data of the gas-phase in the chemical vapor deposition of the Si-B-C-N-H-Cl system include the heat capacities, entropies, enthalpies of formation and Gibbs free energies of formation ware calculated with the reliable theoretical method of quantum mechanics combined with standard statistical thermodynamics. This work provides more fundamental data for analyzing the thermochemistry of the CVD process of the Si-B-C-N-H-Cl system and its subsystems.
SiBCN ceramic; CVD; Thermodynamic
O642
1005-1198 (2016) 04-0280-10
A
10.16253/j.cnki.37-1226/tq.2016.06.005
2016-06-15
2016-06-18
国家自然科学基金 (50572089, 50642039)。
任海涛 (1982 -), 男, 河北保定人, 博士后。E-mail: renht0929@163.com。
