流动注射毛细管电泳测定益母草中4种黄酮
2016-09-29张静姝余丽丽
张静姝,余丽丽
(西安医学院 药学院, 陕西 西安 710021)
·化学与化学工程·
流动注射毛细管电泳测定益母草中4种黄酮
张静姝,余丽丽
(西安医学院 药学院, 陕西 西安710021)
建立了一种测定芦丁、山奈酚、芹菜素和槲皮素的流动注射-毛细管电泳新方法。考察了缓冲溶液种类、缓冲溶液pH、缓冲溶液浓度、有机添加剂含量、分离电压、载液流速、充样时间和注射时间等因素对分离的影响。在优化的实验条件下,4种分析物在9 min内完全分离。回归方程线性良好,相关系数为0.999 0~0.999 9,检出限为1.95~2.59 μg/mL,迁移时间和峰面积的日内及日间相对标准偏差分别小于1.64%和4.16%。用于实际样品益母草中芦丁、山奈酚、芹菜素和槲皮素的测定,回收率为95%~104.3%。
毛细管电泳;流动注射;益母草;黄酮
益母草为唇形科植物益母草(Leonurusjaponicus)的全草,其干燥地上部分为常用中药,具有清热解毒、活血调经、利尿消肿等功效[1]。此外,益母草还具有抑菌、抗血栓、改善冠脉循环、保护心血管等作用,临床上主要用于月经不调、瘀血腹痛、痈肿疮疡和心血管等疾病的治疗[2]。益母草的化学成分主要为生物碱、黄酮、二萜、有机酸及糖类[1],黄酮类化合物具有防癌、抗菌、抗肿瘤、抗氧化等生理活性[3],分析益母草中的黄酮类化合物具有重要意义。
益母草中黄酮类化合物的测定方法包括分光光度法、高效液相色谱法和毛细管电泳法[1,4-5]。分光光度法简单易行,但选择性较差,易受杂质干扰[1];高效液相色谱法灵敏度高、准确性好,但分析周期较长且色谱柱易污染[6];毛细管电泳法具有高效、快速、样品及试剂消耗少等优点,近年来在中草药分析中备受关注[7-8]。然而,传统间歇的进样方式影响了毛细管电泳的分析速度和重现性,限制了其应用。流动注射-毛细管电泳(FI-CE)可在持续高压下连续进样分析,有效提高了传统电泳的分析速度和分析结果的准确性[9-11],是一种高通量的实验技术。本文采用简单、高效的FI-CE法分析芦丁、山奈酚、芹菜素和槲皮素,并将其用于益母草中4种黄酮的含量测定。
1 实验部分
1.1仪器与试剂
HPE-100型电泳仪(美国Bio-Rad公司);未涂层石英毛细管(30.0 cm×75 μm,有效长度26.7 cm,河北永年光导纤维厂);K-1000 FIA型流动注射分析仪(日本HITACHI公司);pHS-3B型酸度计(上海精密科学仪器有限公司)。
芦丁、山奈酚、芹菜素、槲皮素(>98%,陕西慧科植物开发有限公司);益母草样品(兰州盘旋路药房,甘肃兰州);实验所用试剂均为分析纯;实验用水为蒸馏水;实验所用溶液均经0.45 μm微孔滤膜过滤并超声脱气。
新毛细管依次用水(10 min),0.1 mol/L NaOH(30 min),水(10 min)冲洗;测定间隔依次用水、0.1 mol/L NaOH、水、运行缓冲液冲洗3,5,3,3 min;阳极进样,阴极检测;检测波长214 nm。
1.2标准及样品溶液配制
准确称取芦丁、山奈酚、芹菜素、槲皮素标准品,用乙醇溶解并定容配成1.0mg/mL储备液,4℃保存备用,使用时用乙醇稀释至相应浓度进行电泳分析。称取5.0 g研碎的益母草样品于250 mL圆底烧瓶中,加入100 mL乙醇,微沸提取1 h。提取液冷却后过滤,滤液经减压蒸馏浓缩后用乙醇定容至10 mL,4℃保存备用。
2 结果与讨论
2.1分离条件
2.1.1缓冲溶液种类运行缓冲溶液对电渗流、分析物的迁移时间和分离效率有显著影响。实验考察了毛细管电泳中常用缓冲体系(硼砂体系、磷酸盐体系、硼砂—NaH2PO4体系)对分离的影响。结果表明,使用硼砂—NaH2PO4体系时分析物的分离效果最好,因此选择硼砂—NaH2PO4溶液作为运行缓冲溶液。
2.1.2缓冲溶液pH缓冲溶液pH能够影响电渗流及分析物的解离度,从而影响分离选择性和灵敏度。实验在pH 8.0~9.5范围内考察了缓冲溶液pH对分离效果的影响。结果表明,缓冲溶液pH<8.9时,4种分析物未能完全分离;缓冲溶液pH>8.9时,出峰时间变长、峰形变差且灵敏度降低。综合考虑,分离度、分析时间和灵敏度,选择pH 8.9为缓冲溶液最佳pH。
2.1.3硼砂浓度缓冲溶液浓度影响分离体系的离子强度,从而影响电渗流及分离效率。实验保持硼砂和NaH2PO4的浓度比为1∶2(mmol/L),在5~10 mmol/L范围内研究了硼砂浓度对分离的影响。结果发现,分离度和迁移时间均随硼砂浓度的增加而增加。当硼砂浓度为8 mmol/L时,标准混合液及样品中的分析物均能与其他组分完全分离;增大硼砂浓度,随之增加的焦耳热使得分离度减小,灵敏度降低。综合考虑分析时间、分离度和灵敏度,8 mmol/L为硼砂的最佳浓度。
2.1.4有机添加剂缓冲溶液中加入有机添加剂可改变分离介质的离子强度及黏度,从而改变分离效率。实验在0~15%(体积分数)范围内研究了CH3OH含量对分离的影响。结果表明,随着CH3OH含量的增加,分析物出峰时间变长、分离度增大。CH3OH含量为10%(体积分数)时,样品中的分析物可与其他组分完全分离;继续增大CH3OH含量,峰形变差,灵敏度降低。综合考虑分析时间和分离度,选择10%(体积分数)为CH3OH的最佳含量。
2.1.5分离电压毛细管电泳中,分离电压是分离效率的重要影响因素。实验在8.0~10.0 kV范围内考察了分离电压的影响。结果发现,随着分离电压的增大,迁移时间及分离度均减小,当分离电压大于9.5 kV时,样品中的待测物未能与其他组分实现基线分离。综合考虑分析时间及分离效率,9.5 kV被选择为最佳分离电压。
2.1.6充样时间和注射时间实验选择体积为20 μL的样品环及试剂环,考察了充样时间在1~5 s,注射时间在3~11 s范围内变化对分离效果的影响。结果表明,样品环及试剂环只需3 s即可充满,故选择3 s为最佳充样时间;随着注射时间的增加,灵敏度不断提高,当注射时间超过9 s时,峰形展宽,分离度降低,故选择9 s为最佳注射时间。
2.1.7载液流速在FI-CE体系中,载液流速决定了样品区带通过毛细管口的时间,对分离选择性和灵敏度具有显著影响[12]。实验在0.6~1.4 mL/min范围内考察了载液流速对分离的影响。结果显示,灵敏度随载液流速的增大先升高后降低,流速较低时,峰形展宽。综合考虑,灵敏度和峰形,选择1.0 mL/min为最佳载液流速。
2.2FI-CE体系分析性能
2.2.1标准曲线和检出限在优化的实验条件下,芦丁、山奈酚、芹菜素和槲皮素的回归方程、相关系数、线性范围及检出限如表1所示。结果表明,回归方程线性良好,检出限(S/N=3)为1.95~2.59 μg/mL。
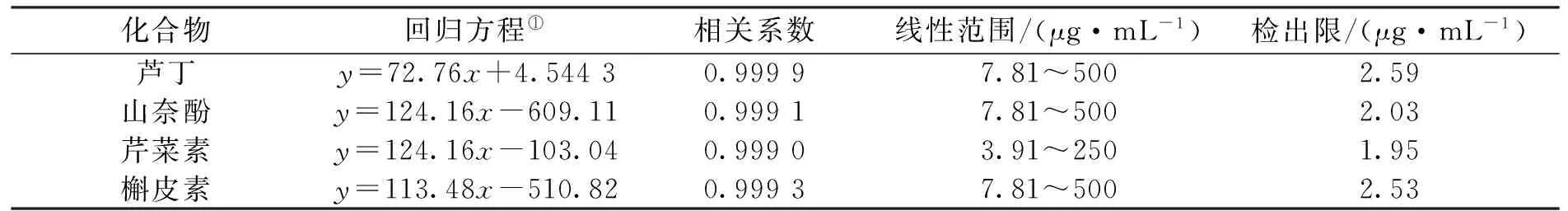
表1 4种黄酮的回归方程、相关系数、线性范围及检出限
注:①y为分析物峰面积(mV·s);x为分析物浓度(μg/mL)。
2.2.2精密度在最佳条件下,将250 μg/mL芦丁、山奈酚、芹菜素和槲皮素的标准混合溶液每隔4min进样,重复进样6次(进样频率为15 h-1),电泳谱图如图1所示。用迁移时间和峰面积的日内及日间相对标准偏差(RSD)对方法的精密度进行考查,结果如表2所示,4种化合物的RSD均小于4.16%。
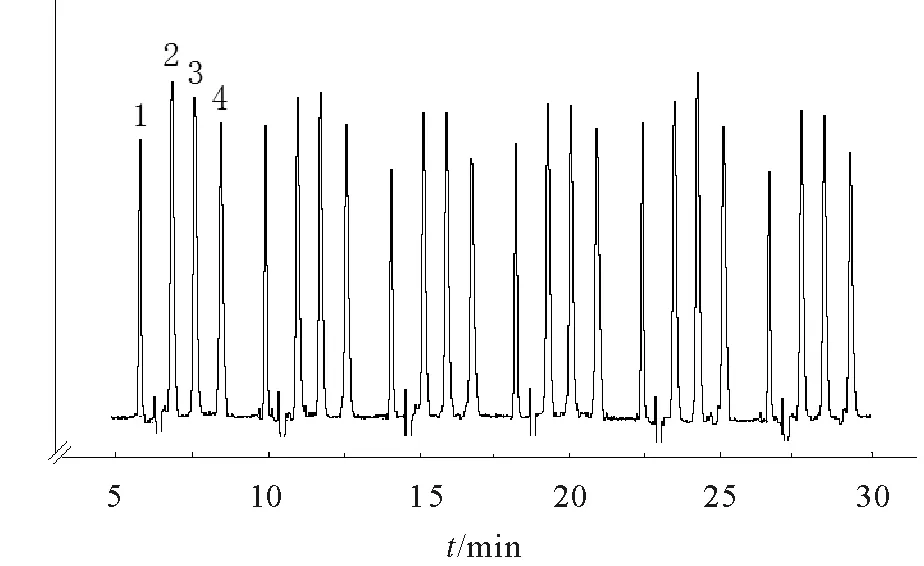
1. 芦丁;2. 山奈酚;3. 芹菜素;4. 槲皮素条件:熔融石英毛细管(30.0 cm×75 μm,有效长度26.7 cm);8 mmol/L 硼砂-16 mmol/L NaH2PO4-10%(V/V)CH3OH(pH 8.9)缓冲体系;分离电压:9.5 kV;检测波长:214 nm;充样时间:3 s;注射时间:9 s;载液流速:1.0 mL/min。图1 标准混合溶液连续6次进样电泳谱图Fig.1 Electrochromatogram of standard mixed solution through six consecutive sampling

化合物RSD/% 迁移时间 峰面积 日内日间日内日间芦丁0.281.641.991.53山奈酚0.221.074.103.74芹菜素0.160.964.161.05槲皮素0.181.443.282.22
2.3样品分析
为考察方法的实用性,用所建立的FI—CE方法对实际样品益母草中的芦丁、山奈酚、芹菜素和槲皮素的含量进行测定,益母草的电泳谱图和实验分析结果分别如图2及表3所示。结果表明,4种分析物的回收率为95%~104.3%,RSD均小于2.8%。
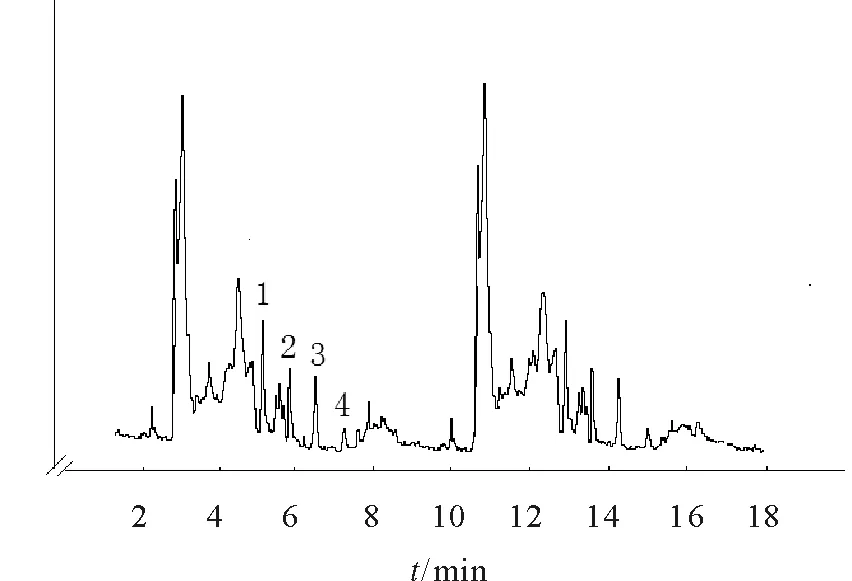
1. 芦丁;2. 山奈酚;3. 芹菜素;4. 槲皮素条件:同图1所述。图2 益母草电泳谱图Fig.2 Electropherogram of Leonurus japonicus
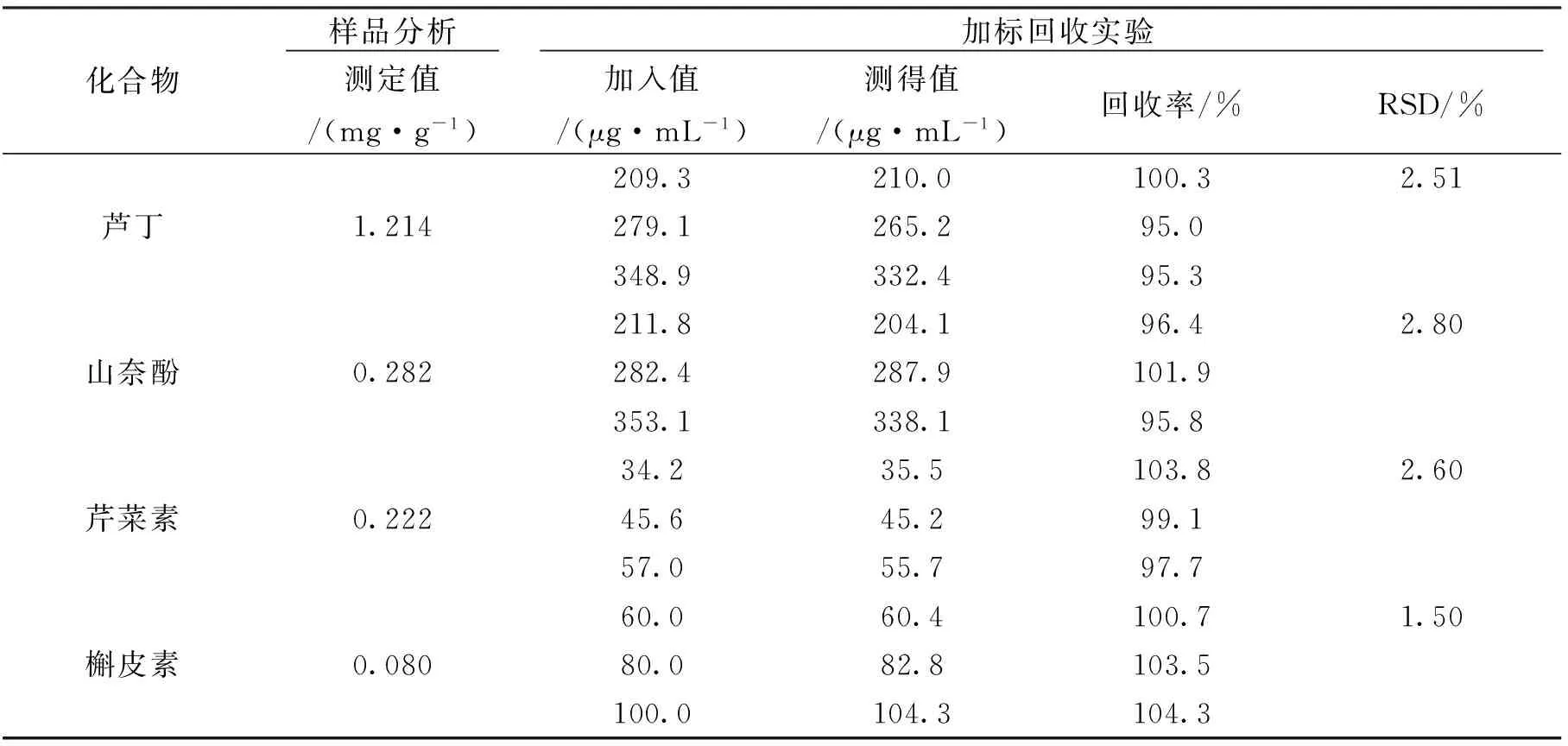
化合物 样品分析 加标回收实验 测定值/(mg·g-1)加入值/(μg·mL-1)测得值/(μg·mL-1)回收率/%RSD/%209.3210.0100.32.51芦丁1.214279.1265.295.0348.9332.495.3211.8204.196.42.80山奈酚0.282282.4287.9101.9353.1338.195.834.235.5103.82.60芹菜素0.22245.645.299.157.055.797.760.060.4100.71.50槲皮素0.08080.082.8103.5100.0104.3104.3
3 结 论
本文建立了一种测定益母草中芦丁、山奈酚、芹菜素和槲皮素等4种黄酮类化合物的FI-CE新方法。此方法可在不间断高压的情况下连续进样,有效缩短分析时间并提高方法的精密度和准确度,具有简单、高效等特点,可用于天然药物中黄酮类化合物的含量测定及相关药物的质量监控。
[1]翁爱彬,郑荔莉,方剑英.HPLC法测定益母草中的黄酮类化合物[J].河北医药,2013,35(6):926-927.
[2]魏丽春,李庆军.益母草的药理与临床研究进展[J].西北药学杂志,2009,24(4):333-335.
[3]延玺,刘会青,邹永青,等.黄酮类化合物生理活性及合成研究进展[J].有机化学, 2008,28(9):1534-1544.
[4]吴婷,管月清,郑双杰,等.毛细管电泳-电化学检测益母草及其冲剂中的黄酮类化合物[J].分析科学学报,2006,22(4):406-409.
[5]黄锁义,黎海妮,余美料,等.益母草总黄酮的提取及鉴别[J].时珍国医国药,2005,16(5):398-399.
[6]张礼春,曾凯,高舸,等.高效毛细管电泳法同时测定饮料中七种防腐剂[J].分析实验室,2015,34(1):77-80.
[7]王彩霞, 闫宏涛, 张全彩.高效毛细管电泳法测定牛奶和奶糖中的三聚氰胺[J].西北大学学报(自然科学版),2009,39(4):599-602.
[8]王敦青,贾春晓,孔春燕,等.极性转换大体积样品堆积-区带毛细管电泳法分析山奈酚、木犀草素和槲皮素[J].化学试剂,2015,37(2):149-151;188.
[9]KUBAN P, KUBAN P, KUBAN V. Flow injection-capillary electrophoresis system with contactless conductivity detection and hydrostatic pressure generated flow. Application to the quantitative analysis of inorganic anions in water samples [J].Electrophoresis, 2003, 24(12-13):1935-1943.
[10] FAN Liu-yin, CHEN Hong-li, CHEN Xing-guo, et al. Separation and determination of sulfonamides inpharmaceutical preparations by a microfluidic capillary electrophoresis system with a continuous sample introduction interface [J].J Spe Sci, 2003, 26(15-16):1376-1382.
[11] LIU Li-hong, FAN Liu-yin, CHEN Hong-li, et al. Separation and determination of four active anthraquinones in Chinese herbal preparations by flow injection-capillary electrophoresis [J].Electrophoresis, 2005, 26(15):2999-3006.
[12] FANG Zhao-lun, LIU Zhi-song, SHEN Qi. Combination of flow injection with capillary electrophoresis. Part I. The basic system [J].Anal Chim Acta, 1997, 346(2):135-143.
(编辑陈镱文)
Determination of four flavonoids inLeonurusjaponicasby flow injection-capillary electrophoresis
ZHANG Jing-shu, YU Li-li
(College of pharmacy, Xi′an Medical University, Xi′an 710021, China)
A new method was developed based on flow injection-capillary electrophoresis technique to determine rutin, kaempferol, apigenin and quercetin. Multiple factors affecting separation such as the running buffer type, buffer pH, buffer concentration, the content of organic additive, separation voltage, the flow-rate of carrier solution, charging time and injecting time have been investigated. Four analytes were well separated from each other in 9 min under optimal conditions. The regression equations have excellent linearity. Correlation coefficients ranged from 0.999 0 to 0.999 8 and limits of detection ranged from 1.95 to 2.59 μg/mL. The relative standard deviations of migration time and peak area for intra-day and inter-day analysis were less than 1.64% and 4.16%, respectively. Applied to determine rutin, kaempferol, apigenin and quercetin inLeonurusjaponicasreal sample and the recoveries obtained were in the range of 95%~104.3%.
capillary electrophoresis; flow injection;Leonurusjaponicus; flavonoids
2015-11-10
国家自然科学基金资助项目(81302706)
张静姝,女,陕西靖边人,从事分析化学研究。
O657.8
A
10.16152/j.cnki.xdxbzr.2016-03-014
