2015年四川省麻疹病毒分子流行病学特征分析
2016-09-28曹冉冉刘李朱昱锟王顺东何吉兰
曹冉冉 刘李 朱昱锟 王顺东 何吉兰
610041 成都,四川省疾病预防控制中心(曹冉冉、刘李、何吉兰);637000 南充市疾病预防控制中心(朱昱锟);635000 达州市疾病预防控制中心(王顺东)
·论著·
2015年四川省麻疹病毒分子流行病学特征分析
曹冉冉刘李朱昱锟王顺东何吉兰
610041 成都,四川省疾病预防控制中心(曹冉冉、刘李、何吉兰);637000 南充市疾病预防控制中心(朱昱锟);635000 达州市疾病预防控制中心(王顺东)
目的 分析四川省2015年流行的麻疹病毒的基因型别和核蛋白基因特征,了解不同基因型别麻疹病毒在省内流行情况。方法采集疑似麻疹病例咽拭子标本,用Vero/SLAM细胞分离病毒。对分离培养阳性毒株,采用逆转录-聚合酶链反应(RT-PCR)扩增核蛋白N基因片段并进行序列测定和分析。结果从971份疑似麻疹病例咽拭子标本中分离得到398株麻疹病毒。397株分离株为H1基因型,与H1基因型参考株核苷酸同源性为96.2%-98.0%;在进化树上呈现为2个分支,分支间平均核苷酸差异率为2.0%。1株分离株为A基因型,与A基因型参考株核苷酸同源性为98.0%。结论H1基因型为2015年四川省本土流行麻疹病毒的优势基因型;近年来四川省内流行的毒株未发生较大变化,但存在一定核苷酸差异,呈现2个传播链,需进一步加强监测。
【主题词】麻疹病毒;核蛋白类;基因型
麻疹是常见的急性呼吸系统传染病,传染性极强,易引起暴发流行。随着天花的消灭以及无脊灰状态在多数地区的实现,麻疹被WHO列为下一个拟被消除的传染病。但2005年我国大部分地区麻疹疫情有所回升[1-3]。四川省从2009年以来麻疹疫情也有所上升,局部地区发生麻疹暴发流行[4]。特别是2015以来,仅上半年疑似麻疹病例数达971例,远超2010-2014年同期平均水平74例。为从分子水平分析四川省麻疹流行的原因,探讨消除麻疹策略,对四川省2015年上半年分离到的398株麻疹病毒核蛋白N基因进行序列测定,并进行基因型别鉴定和特征分析。
1 材料与方法
1.1标本采集采集疑似麻疹病例咽拭子标本,保存在病毒标本运输液中,-20 ℃或以下温度保存,冷冻运输。标本用终浓度为1 000 U /ml的青霉素和1 000 g /ml的链霉素处理,4 ℃保存备用。
1.2病毒分离将处理后的标本接种于生长良好的 Vero /SLAM 单层细胞,置5% CO2培养箱,37 ℃连续培养7 d,每日观察细胞病变。当75% 以上的细胞呈现特异性巨细胞融合病变时收获。若无病变需连续盲传 3代,仍无细胞病变则为阴性。
1.3病毒RNA提取和逆转录-聚合酶链反应(RT-PCR)用Promega公司Maxwell 16 Viral Total Nucleic Acid Purification Kit按试剂盒说明书提取病毒 RNA。用Invitrogen公司的Super Script III One-step RT-PCR System with platinumTaqDNA Polymersase试剂盒进行RT-PCR反应,扩增编码N蛋白羧基末端的核苷酸序列。引物MV216:5′- TGGAGCTATGCCATGGGAGT-3′(1 104-1 123 bp),引物MV214:5′-TAACAATGATGGAGGGTAGG-3′(1 737-1 718 bp),扩增产物长度为634 bp。PCR扩增条件:50 ℃ 25 min,94 ℃2 min; 94 ℃15 s、55 ℃ 30 s、68 ℃75 s,40个循环; 68 ℃5 min。扩增产物经琼脂糖凝胶电泳鉴定。
1.4序列测定扩增产物送成都擎科梓熙生物有限技术公司进行序列测定。
1.5序列分析核苷酸序列经Sequencher 5.0校对后,与各基因型参考株核苷酸序列进行比对(编码N蛋白羧基末端的456个核苷酸序列),用MEGA 4.1软件作进化树分析(Kimura 2参数模型,邻位-连接法,1 000次bootstrap检验),根据分支聚集确定基因型别[5]。
2 结果
2.1病毒分离四川省下设21个市州,2015年上半年共采集971份疑似麻疹病例咽拭子标本,来源于省内17个市州。绵阳市、遂宁市、自贡市和内江市无报告病例,因此未采集标本。标本接种Vero /SLAM细胞后,分离得到398株麻疹病毒。
2.2RT-PCR398株麻疹病毒分离物经RT-PCR扩增,均出现大小为634 bp的特异性条带。

图1 2015年四川省398株麻疹病毒与各基因型代表株进化关系树Fig.1 Phylogenetic tree of 398 measles virus isolates in Sichuan province in 2015 compared with Reference Strains for each genotype
2.3序列测定和分析2015年四川省398株麻疹病毒与各基因型代表株共同建立进化关系树(图1)。397株分离株与H1基因型参考株(AF045212)聚集在同一分支上,核苷酸同源性为96.2%-98.0%,属于H1基因型;与H1a基因亚型参考株(Chin9322)核苷酸同源性为97.3%-99.1%,属于H1a基因亚型;397株在系统进化树上呈现2个分支,命名为Cluster Ⅰ和Cluster Ⅱ,分支间平均核苷酸差异率为2.0%。1株分离株与A基因型参考株(U01987)聚集在同一分支上,核苷酸同源为98.0%,与疫苗株Changchun47、Shanghai191同源性分别为97.8%、98.0%。
2.4麻疹病毒ClusterⅠ和Cluster Ⅱ在四川省内分布情况各市州采集标本数、麻疹病毒Cluster I和Cluster II毒株数及构成比见表1,Cluster I和Cluster II在四川地理分布见图2。

图2 麻疹病毒Cluster I和Cluster II在四川地理分布Fig.2 Geographic distribution of measles virus Cluster I and II in Sichuan Province
2.5四川省内毒株变异情况将2011年、2014、2015年分别分离到的10株、86株、398株麻疹病毒序列进行进化树分析。结果显示,492株为H1a基因亚型,毒株间核苷酸同源性为96.2%-100.0%,在进化树上仍呈现两个分支;2株为A基因型,毒株间核苷酸同源性为99.7%。
3 讨论
麻疹病毒是副粘病毒科麻疹病毒属的单股负链RNA病毒,有6个结构基因,分别编码核蛋白(N)、磷酸蛋白(P)、膜蛋白(M)、融合蛋白(F)、血凝素蛋白(H)和依赖于RNA的RNA聚合酶(L)。H基因和N基因是结构基因中易变异的两个基因,世界卫生组织规定编码N蛋白羧基末端450 bp核苷酸序列或H基因全长编码序列作为麻疹病毒基因定型依据。研究参考中国CDC提供引物序列,对四川省2015年上半年分离到的398株麻疹病毒编码N蛋白羧基末端的456 bp核苷酸片段进行扩增、测序和分析。结果显示,397株分离株与H1a基因亚型代表株核苷酸同源性为97.3%-99.1%,为H1a基因亚型,与全国优势流行株的基因型别一致[6-11]。397株分离株之间核苷酸同源性为96.2%-100.0%,虽变异程度小,但在系统进化树上呈现2个分支(命名为Cluster I和Cluster II),分支间平均核苷酸差异率为2.0%,提示在四川省内麻疹病毒的流行和传播存在两个传播链。
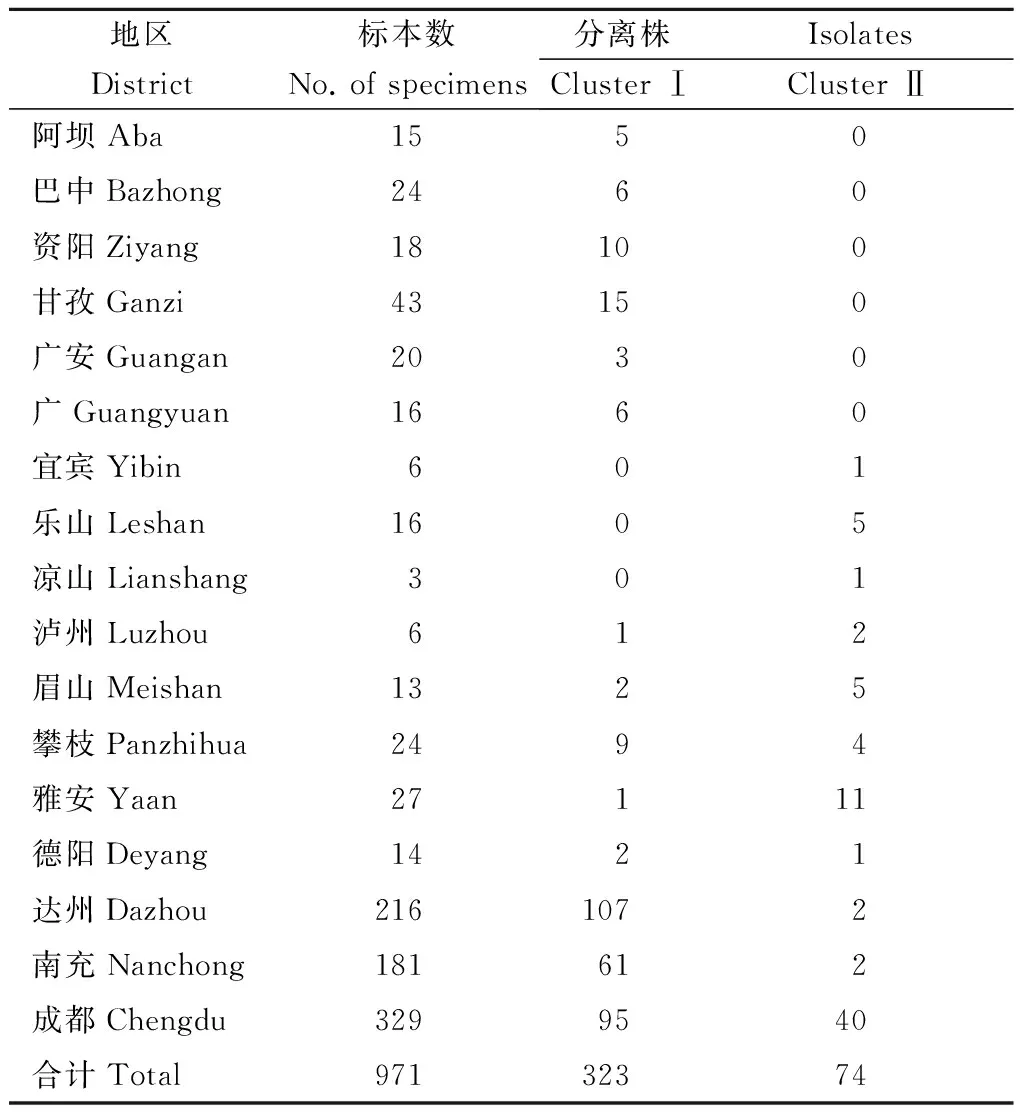
表1 各市州采集标本数和麻疹病毒Cluster I
四川省下设21个市州,397株分离株来源于17个市州(4个市州无报告病例,未采集标本),Cluster I、II所占比例分别为81.4%(323/397)、18.6%(74/397)。从覆盖区域来看,6个市州仅有Cluster I流行,主要分布在四川省西北部;3个市州仅有Cluster II流行,8个市州Cluster I、II同时流行,主要分布在四川省东南部。因此,从毒株数量和覆盖范围来看,Cluster I都是2015年四川省麻疹流行的主要传播链。
2015年分离到1株A基因型麻疹毒株(2015-727)。病例与麻风腮疫苗接种时间、出疹时间都非常接近,高度怀疑为疫苗相关株。通过比对,2015-727与疫苗株Changchun47、Shanghai191同源性非常高,表明四川省分离到疫苗相关株保持高度稳定。
病毒学监测是消除麻疹的重要手段之一。通过对麻疹病毒核酸序列的分析,可及时了解和掌握麻疹流行株的基因型别和特征,追踪传染源和传播途径,为科学防麻疹提供参考。
[1]耿倩,陈蓉,张涛,等. 2006-2011年上海市麻疹流行病学特征分析[J]. 中华疾病控制杂志,2013,17(11): 955-958.
[2]张大勇,戴丽芳,徐飞,等. 贵州2003-2010年麻疹流行病学特征及消除麻疹策略分析[J]. 中华疾病控制杂志,2013,17(2):163-166.
[3]方巧云,曾健君,刘燕,等. 惠州市2004-2010年麻疹流行病学特征分析[J]. 中华疾病控制杂志,2012,16(3):241-243.
[4]刘李, 孙莉, 何吉兰,等. 2011年上半年四川省麻疹病毒分离株N基因序列分析[J]. 预防医学情报杂志, 2012, 28(6):426-429.
[5]童文彬, 何吉兰, 孙莉,等. 四川省1株输入性麻疹病毒基因特征分析[J]. 预防医学情报杂志, 2009, 25(11):927-930.
[6]Zhang Y, Deng ZR, Wang HL, et al. New measles virus genotype associated with outbreak, China.[J]. Emerg Infect Dis, 2010, 16(6):943-947. doi: 10.3201/eid1606.100089
[7]陈超, 周剑惠, 张帆,等. 吉林省2009-2010年麻疹病毒流行株核蛋白基因亲缘关系及氨基酸变异分析[J]. 中国疫苗和免疫, 2012,18(1):43-47.
[8]许青, 李波, 房学强,等. 山东省首起境外输入性D9基因型麻疹病毒引发的暴发疫情调查[J]. 中华预防医学杂志, 2014, 48(7):638-639. doi:10.3760/cma.j.issn.0253-9624.2014.07.021
[9]雷亚克, 戴莹, 李静,等. 2008-2012年湖北省麻疹野病毒分子流行病学研究[J]. 中华实验和临床病毒学杂志, 2015, 29(4):303-305. doi:10.3760/cma.j.issn.1003-9279.2015.04.005
[10]王常银, 许松涛, 熊萍,等. 山东省2005-2008年麻疹病毒流行特征分析[J]. 中国疫苗和免疫, 2011,17(1):22-25.
[11]Zhang Y, Xu ST, Wang HL, et al. Single Endemic Genotype of Measles Virus Continuously Circulating in China for at Least 16 Years[J]. PloS One, 2012, 7(4): e34401. doi:10.1371/journal.pone.0034401
Molecular epidemiological characteristics of measles virus in Sichuan province in 2015
CaoRanran,LiuLi,ZhuYukun,WangShundong,HeJilan
SichuanProvincialCenterforDiseaseControlandPrevention,Chengdu610041,China(CaoRR,LiuL,HeJL);NanchongCenterforDiseaseControlandPrevention,Nanchong63700,China(ZhuYK);DazhouCenterforDiseaseControlandPrevention,Dazhou63500,China(WangSD)Correspondingauthor:HeJilan,Email:275880976@qq.com
ObjectiveTo analyze the genetic characteristics of measles virus in Sichuan province in 2015. MethodsMeasles virus was isolated from throat swab specimens collected from suspected measles cases. Nucleotide sequences coding for C terminus of nucleoprotein were amplified by RT-PCR and sequenced for phylogenetic analysis. Results398 measles virus isolates were obtained from 971 throat swab specimens. Phylogenetic analysis showed that 397 belonged to H1 genotype, the sequences homologies were 96.2%-98.0% compared with H1 genotype reference strain; and all those isolates separated into 2 clusters in phylogenetic tree with the average nucleotide divergence of 2.0%.1 isolate belonged to A genotype, and shared 98.0% nucleotide homology compared with A genotype reference strain. ConclusionsH1 genotype virus remained predominant circulating in Sichuan Province. There was no much genetic variation in N gene. But a minor nucleotide divergence existed between different clusters, leading to 2 transmission chains.
Measles virus; Nucleoproteins; Genotype
何吉兰,Email: 275880976@qq.com
10.3760/cma.j.issn.1003-9279.2016.04.010
2016-01-15)
