油菜开花期QTL定位及与粒重的遗传关联性
2016-09-09黄吉祥熊化鑫倪西源张晓玉赵坚义
黄吉祥,熊化鑫,2,潘 兵,3,倪西源,张晓玉,赵坚义
(1浙江省农业科学院作物与核技术利用研究所/植物有害生物防控国家重点实验室省部共建培育基地,杭州 310021;2浙江师范大学化学与生命科学学院,浙江金华 321000;3浙江大学农业与生物技术学院,杭州 310058)
油菜开花期QTL定位及与粒重的遗传关联性
黄吉祥1,熊化鑫1,2,潘 兵1,3,倪西源1,张晓玉1,赵坚义1
(1浙江省农业科学院作物与核技术利用研究所/植物有害生物防控国家重点实验室省部共建培育基地,杭州 310021;2浙江师范大学化学与生命科学学院,浙江金华 321000;3浙江大学农业与生物技术学院,杭州 310058)
【目的】明确中国和欧洲油菜开花期主控位点及其对粒重的影响,为早熟油菜品种选育提供科学依据。【方法】以欧洲冬油菜Sollux和中国品种高油605的选系(Gaoyou)杂交F1经小孢子培养产生的DH群体为材料,采用7年9种环境下的开花期表型数据和新版SG图谱定位开花期QTL,并采用条件遗传学和QTL分析相结合的条件QTL定位方法,解析开花期对千粒重QTL的影响,最后对各20个极端开花期株系的基因型和表现型进行性状-标记的符合度测定,为标记筛选用于辅助选育提供依据。【结果】应用WinQTLCart 2.5复合区间作图法,共检测到7个在3种以上环境中稳定表达的控制开花QTL,加性效应值在0.58—3.85 d,解释了表型总变异的84%。8对上位性QTL效应总和为加性总效应的41.8%。QTL与环境互作效应只在少数位点和个别环境中显著。在3个主效QTL峰值或相近位置上定位了4个在拟南芥中调控开花的关键基因FT、API、FLC和FY的6个同源拷贝,为发掘控制这些QTL的候选基因提供了有价值的参考信息。条件QTL分析表明,在4个增重效应均来自Gaoyou的千粒重QTL位点(qSWA2、qSWA3、qSWA4和qSWC2),大粒等位基因效应可能与开花早、籽粒灌浆期长有关。通过选择这些位点的早开花标记基因型有望同时提高种子千粒重,这也部分给出了开花期与千粒重之间极显著负相关的遗传解释,但2个粒重主效位点(qSWA7和qSWC8)的遗传效应不受开花期影响。根据SG群体极端开花期株系在3个效应值最大的QTL(qFTA2、qFTC2和qFTC6)区域标记基因型和开花期表现型的关联分析,筛选获得6个高质量、高吻合度的共显性标记推荐育种应用。qFTA2位点,标记辅助准确率为70%-80%;qFTC2 和qFTC6位点的选择效率达到80%-100%。基因型组配分析显示,聚合qFTA2、qFTC2和qFTC6的早开花等位基因,可显著提早开花期,同步增加千粒重但不影响含油量和角果粒数。【结论】7个QTL均显示早开花等位基因来自中国亲本。拟南芥中调控开花关键基因FT、API、FLC和FY的6个同源拷贝定位到3个主效QTL峰值位置。开花迟、早显著影响4个千粒重QTL位点,但2个最重要的粒重位点(qSWA7和qSWC8)不受影响;3个主效QTL(qFTA2、qFTC2和qFTC6)的6个共显性标记可用于早熟基因的转育和早熟材料的筛选。
甘蓝型油菜;QTL定位;开花期;千粒重;条件QTL定位
0 引言
【研究意义】油菜开花期与籽粒成熟期高度相关[1],育种中通常将其作为选择早熟性的重要指标[2]。开花时间是植物在自然演化过程中对当地环境的一种适应,关于油菜作物开花基因的研究,目前多数仍停留在QTL和生物信息学分析层面。因此,进一步对主效开花期QTL作精细定位,结合拟南芥基因库信息资源、图位克隆基因、发展基因标记或紧密连锁分子标记用于育种研究,为创制新的亲本资源提供依据。【前人研究进展】DETJEN等[3]和DICKSON等[4]通过对芸薹属作物甘蓝和花椰菜开花性状进行遗传分析,估算遗传力、基因效应及基因数目。20世纪90年代后,随着分子生物学理论和技术的发展,从 QTL (quantitative trait loci)水平上对油菜开花期进行深入的解析。1995年,TEUTONICO等[5]首先在白菜LG2 (A2)和LG8(A8)连锁群上定位到2个主效开花期QTL。此后,FERREIRA等[6]以甘蓝型冬油菜和春油菜杂交F1衍生的DH群体为材料,在A9、C2、C6上定位到3个主效QTL,其中A9连锁群上的QTL贡献率达到28%。接着,OSBORN等[7]在此基础上通过添加大量分子标记,重新定位QTL,使得A9上的QTL贡献率由28%增加到46.9%,A9、C2和C6上3个QTL解释群体遗传总变异的 63.5%。BUTRUILLE等[8]1999年利用冬、春油菜间杂交和回交群体,在连锁群A2、A3、A7、A8、A9、C2和C5上定位到开花期QTL,总贡献率达到59%。ZHAO等[1]2005年用德国品种 Sollux和中国油菜 Gaoyou为亲本构建的SG-DH群体,在德国、中国杭州和西安3种环境下定位分析开花期性状,检测到7个主效QTL,分别位于A1、A2、C1、C2、C4、C6和C9上。其中,A1、C2 和C6相同位点上检测到控制成熟期QTL。LONG等[9]于2007年利用欧洲冬油菜Tapidor和中国品种宁油7号构建的TN群体,在中国冬油菜和春油菜区共11种环境下进行定位试验,发现36个SL-QTL(显著),遍布油菜大多数连锁群,其中A10和C6连锁群上获得春、冬油菜环境中效应值最大、贡献率分别达到52% 和26%—52%的主效QTL。除此,蔡长春等[10]在油菜A4和C6上定位到贡献率达到48.0%和20.6%的QTL。近期,XU等[11]利用60K油菜芯片对523个品种进行SNP位点多态性检测,并对开花期性状进行全基因组关联分析,在A2、A4、A10、C3和C5上关联到在多环境中表达的控制开花期QTL。WE等[12]在C2上定位到贡献率达到20%以上的主效开花QTL。NELSON等[13]在温、光控制条件下研究油菜开花性状,在A2、A7和C3上定位到控制开花主效QTL,解释群体遗传变异的57.7%。总之,控制油菜开花迟早的共性基因位点主要分布在A2、A3、A8、A9、A10以及C2 和C6染色体上。在模式植物拟南芥中,开花基因和调控网络的研究很清楚[14-15]。据统计,超过 300个基因参与拟南芥开花的调控网络[16]。近年来,通过基因组DNA序列线性比对和QTL位置的比较分析,在油菜共性开花期主效 QTL峰值或附近位置均搜索到一系列拟南芥同源开花关键基因[7,9,17],作为多倍体的油菜作物,其开花期的控制机制相对拟南芥更为复杂,因而QTL数量更多,互作关系更趋网状结构。迄今为止,油菜 19条连锁群上几乎都检测到控制开花期的QTL。HOU等[18]首次对甘蓝型油菜A10连锁群上的开花期主效QTL进行图为克隆,精细定位至80 kb区间,确定含FLC同源基因,发现上游启动子区域一个621 bp的插入与油菜冬性特征有关。据报道,油菜开花期与产量性状有较大关联,每果粒数与开花迟、早显著正相关,与千粒重显著负相关[19-20],开花期平均相差约4 d即可导致千粒重显著增减[20]。油菜千粒重是产量形成中最重要的构成因素之一,是一个典型的数量性状,19条连锁群上都检测到千粒重QTL,其中分布在A1、A2、A5、A7、C2、C3、C4、C7、C8 和C9上的QTL被多次报道[21-24]。LIU等[25]通过图位克隆,成功获得控制 A9连锁群上主效粒重 QTL的功能基因。【本研究切入点】近20年中,针对油菜开花期性状,已进行了大量的QTL定位研究,通过不同方法和不同研究材料获得一系列共性主效QTL,但从QTL水平上解析开花期对产量构成性状的影响和相互关系则尚未见报道。【拟解决的关键问题】本研究利用添加了372个标记的新版SG图谱[26]和新增6个试验共9种环境的开花期表型数据,重新对SG群体进行开花期定位分析;同时利用条件QTL分析方法研究开花期对千粒重QTL的影响,为早熟油菜品种选育提供科学依据。
1 材料与方法
1.1 研究材料
以先前构建的欧洲油菜 Sollux和中国材料Gaoyou杂交F1产生的282个DH株系为材料[2]。Sollux为典型欧洲冬油菜,而 Gaoyou是浙江大学育成的早熟品种高油605的选系,2个品种开花期差异近一个月。QTL定位图谱包含481个分子标记,覆盖甘蓝型油菜基因组19条连锁群,总长1 948.6 cM,标记间平均距离4.05 cM[27]。
1.2 田间试验
田间试验包括2001年中国杭州、西安以及德国哥廷根的Reinshof 3个试验点[2]以及2004、2005、2007、2008、2009和2013年杭州(浙江省农业科学院试验农场)共9个环境试验结果。试验采用完全随机排列,2次重复。每株系种植2行,行长2.5—3.0 m,行距0.33 m,株距0.12 m。
1.3 开花期记载和千粒重测定
亲本和DH株系的开花期记载以小区25%植株开第一朵花为标准,数据分析时换算成播种期到开花期的天数,取2次重复观察值平均值。千粒重用BS 124S型电子天平称量,随机抽取小区混收种子测定,精确到小数点后3位,每样品随机称量3次,重复间相差不超过0.02 g。
1.4 表型数据处理及条件效应预测
DH群体性状表型统计以及性状间相关系数采用SPSS17.0软件。条件效应值的获取采用ZHU[27]提出的基于混合线性模型的数量性状条件分析方法,YT1/T2表示在消除性状2的表型变异后性状1的表型值,如YSW/FT指剔除开花期影响后千粒重(SW)的表型值。
1.5 QTL定位分析
首先运用WinQTLCart 2.5复合区间作图法[28],对开花期表型值按单环境进行全基因组 QTL扫描,以LOD>2.5作为阀值判断QTL是否真实存在。在每个连锁群上间隔1 cM检测QTL存在的可能性,确定各性状QTL的数目及其连锁图上QTL的置信区间、加性效应值以及对性状表型的贡献率。若不同环境中检测到的QTL置信区间(1-LOD)内有重叠区域,则认为是同一个QTL,重叠部分被定义为这个QTL的置信区间。进一步采用QTLNetwork 2.0软件[29],对9种环境下的表型数据进行联合定位分析,估测QTL与环境的互作以及控制开花期基因位点之间的上位性效应。QTL的显著性概率值为0.05,通过1 000次排列,检验推断QTL及成对推断QTL的效应显著性,用检验出的显著QTL构建QTL全模型,进一步对模型进行选择,剔除可能的假阳性QTL,用基于Gibbs抽样的Bayesian方法[30]估计QTL及其与环境互作的各项遗传效应值。
QTL命名采用q+性状英文缩写开花期FT(千粒重SW)+所在连锁群代号+QTL个数。如在油菜第2染色体上第1个开花期QTL,命名为:qFTA2-1。
1.6 候选基因定位
提取SG-DH群体双亲Sollux和Gaoyou抽苔现蕾期DNA,对其进行二代测序,获得现蕾期转录组数据。根据CHALHOUB等[31]近期发表的油菜基因组信息,针对SG群体控制开花期主效QTL区间标记信息,通过分析比对,找出所有表达基因,将这些基因与NCBI上报道的拟南芥开花相关基因比对(P<0.01),获得相关的候选基因并确定候选基因在连锁群上的物理位置。
2 结果
2.1 亲本及DH群体开花期表型变异
通过对亲本和DH群体开花期表型的统计(表1),2个亲本在9种不同环境下的开花期相差17—30 d,平均相差24 d;DH群体株系播种至开花天数9种环境平均最大值196 d,和Sollux相同,最小值170 d,较Gaoyou早2 d,没有明显的超双亲现象,说明控制迟和早开花的等位基因主要分别来自 Sollux和Gaoyou。

表1 DH群体开花期9种环境下的表型变异Table 1 Phenotypic variation of flowering time in SG-DH population across nine environments
2.2 开花期QTL分析
利用WinQTLCart2.5软件对9种环境下的开花期进行QTL扫描,共检测到在3种以上环境中稳定表达的QTL有7个(表2),分布在9条不同的染色体(qFTA2、qFTA3、qFTA4、qFTA10、qFTC2、qFTC6 和 qFTC8)上,平均加性效应值 0.98—2.93。qFTC2 和 qFTC6在 9种环境中稳定表达,峰值集中,平均LOD值达到17.3和15.4(图1-a—图1-b),平均遗传贡献率分别达19.1%和14.9%。qFTA2也在7种环境中被检测到,平均加性效应值为1.52(图1-c),贡献率为7.9%。qFTA3和qFTA10分别在6和5种环境中显著。所有7个QTL位点,其遗传效应均表现为Sollux等位基因使开花延迟而 Gaoyou等位基因致提早开花。当7个QTL位点分别聚合Sollux和Gaoyou等位基因时,可致开花期相差21.8 d,解释群体内遗传总变异的约84%(DH群体内最迟和最早开花的株系相差26 d)。通过对双亲花蕾转录组测序、基因表达分析和序列比对,在3个主效QTL(qFTA2、qFTC2 和qFTC6)区间,发现与拟南芥中控制开花关键基因FLOWERING LOCUS T(FT)的2个同源拷贝BnFT-A2 和 BnFT-C6;春化途径中关键基因 FLOWERING LOCUS C(FLC)以及与FLC相关的酵母多聚腺苷酸化因子pfs2p的同源基因BnFLC-C2和BnFY-C2;还有 APETALA 1-1(AP1-1)和 AP1-2的同源基因BnAP1-C6a和BnAP1-C6b分别位于SG群体中最重要的3个QTL的峰值或附近位置。根据这些同源基因的油菜序列设计标记,已将这6个基因拷贝定位在相应的QTL峰值或相近位置上(图1)
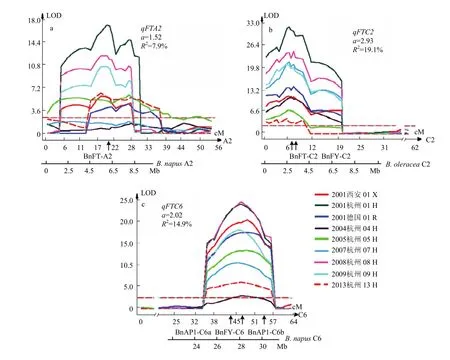
图1 SG-DH群体在9种环境中检测到的3个主效开花期QTL qFTA2(a), qFTC2(b)和qFTC6(c)Fig. 1 Three major QTL of flowering time qFTA2(a), qFTC2(b) and qFTC6(c)from 9 environments in SG DH population
2.3 开花期QTL的上位性及与环境的互作
为解析油菜开花期QTL之间以及QTL与环境之间的互作关系,进一步采用QTLnetwork2.0软件对9种环境下的开花期进行 QTL联合定位,共检测到 8对显著的上位性QTL,5对发生在QTL×QTL之间、2对是QTL×标记位点互作、只有1对是标记位点间互作(表3)。除1对A基因组间和1对C基因组间互作外,其余6对均发生在A和C基因组之间;所有上位性QTL的效应值为负值,说明双亲在这些QTL或标记位点之间杂结合时会相对延迟开花;上位性QTL的效应值在0.32—1.53。总和是加性总效应的41.8%。
结果显示,开花期QTL受环境影响较小,互作(QE)只发生在个别环境的极少数 QTL位点。qFTA4在2001德国和2012年杭州环境中各出现效应值方向相反的 QE互作,效应值分别是 0.8和-0.40。qFTA10在2001的西安和德国环境中存在显著的QE互作,效应值分别为-0.46和0.41,其余QTL未检测到显著的 QE互作,上位性 QTL只在德国Reinshof 环境下,ZAAS1029/qFTC2位点检测到微效的AAE互作。
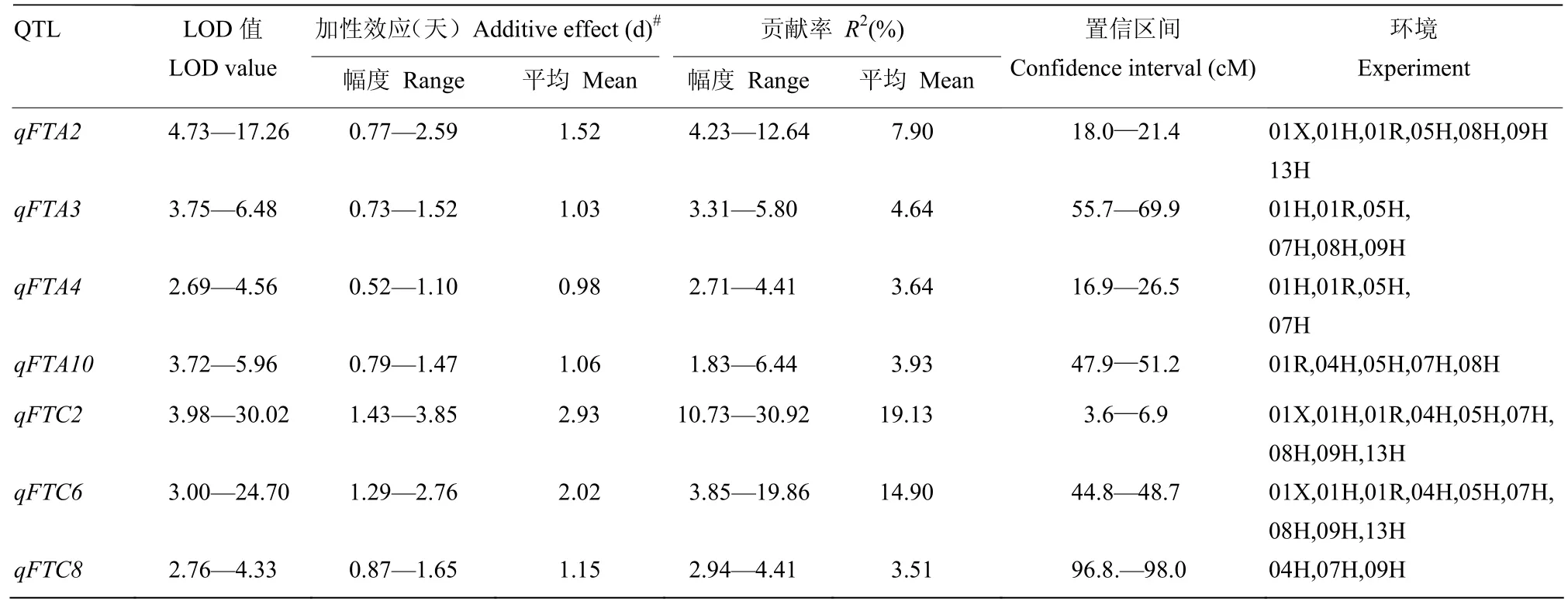
表2 SG-DH群体在9种环境下的开花期QTL信息Table 2 QTL information of flowering time in SG population over 9 environments
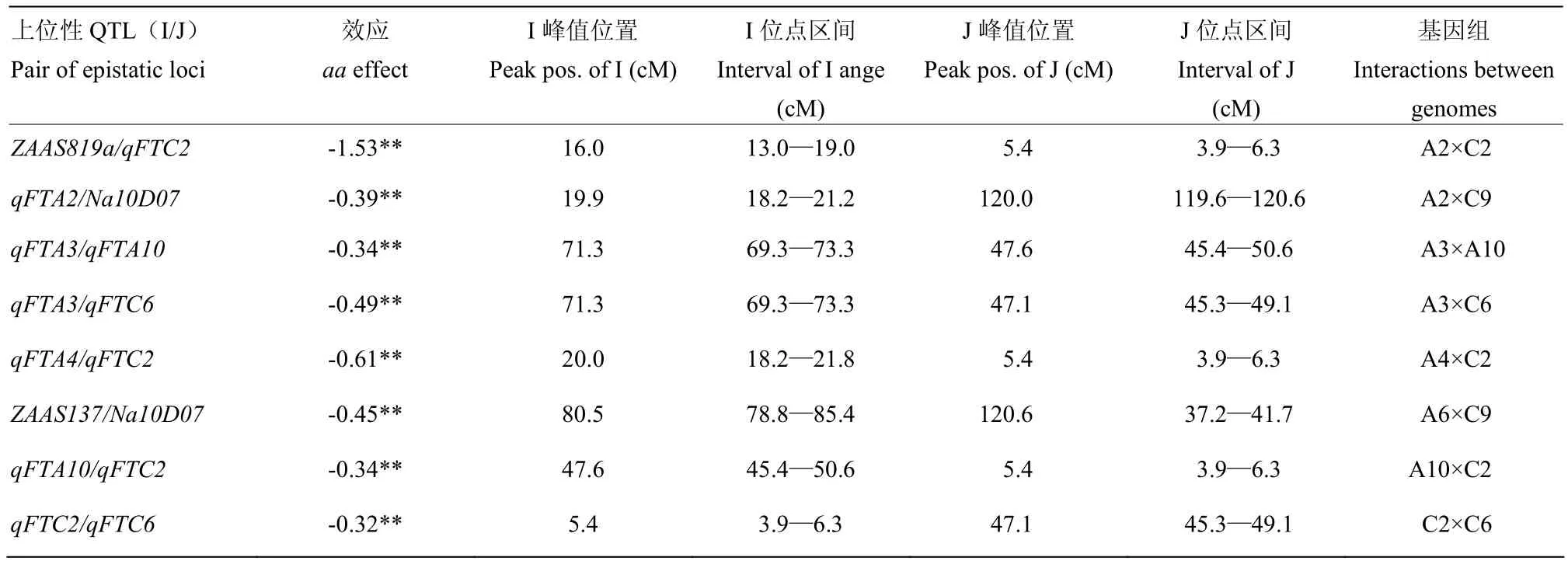
表3 SG-DH群体中开花期的上位性效应Table 3 Epistatic interactions estimated from SG-DH population for flowering time
2.4 开花期与种子千粒重的遗传关联
选用9种环境下SG群体282个株系开花期平均值(播种到开花天数)分别与相应的含油量、千粒重、角果粒数和角果长度均值进行相关分析,结果是开花期与含油量、角果长度和角果粒数的相关系数分别为0.018、0.056和0.008,基本无相关,但和千粒重的相关系数达到-0.403,极显著负相关(P<0.001)。说明开花早的材料可能通过较长的籽粒灌浆周期增加千粒重。为了揭示开花期对千粒重QTL的影响,根据条件QTL定位方法,用9种环境下的平均千粒重和排除开花期变异后的条件千粒重平均值进行条件前、后的QTL分析和比较(表4),当排除开花期条件因素后,在10个千粒重QTL中,6个(qSWA1、qSWA7、qSWC7、qSWC1-2、qSWC1-1和qSWC8)基本保持效应值稳定,除qSWC1-1和qSWC1-2显著性保持不变和略有下降外、其余4个QTL LOD值均有上升。6个QTL位点中,4个增加粒重等位基因来自欧洲亲本 Sollux;值得注意的是,中国亲本Gaoyou等位基因提高粒重的4 个QTL(qSWA2、qSWA3、qSWA4和qSWC2),当给定群体株系间开花期无变化时,均不存在显著的QTL,说明开花期对种子千粒重在这些QTL位点上有显著影响。qSWA7和qSWC8(图2-a—图2-b)是2个显著性最高效应值最大的粒重QTL,显然开花迟、早对其未发生大的影响;qSWC2大粒等位基因来自Gaoyou,是受到开花期调控的典型基因位点,假定以开花期无变化为条件时,LOD值由条件前的5.27降至1左右,qSWC2SW|FT效应不显著(图2-c)。开花期对qSWC7的影响则表现另一种调控模式,非条件qSWC7 的LOD值刚过阀值(2.70),但条件QTL qSWC7SW|FT的LOD值升至3.81,效应值也略增大(图2-d)。
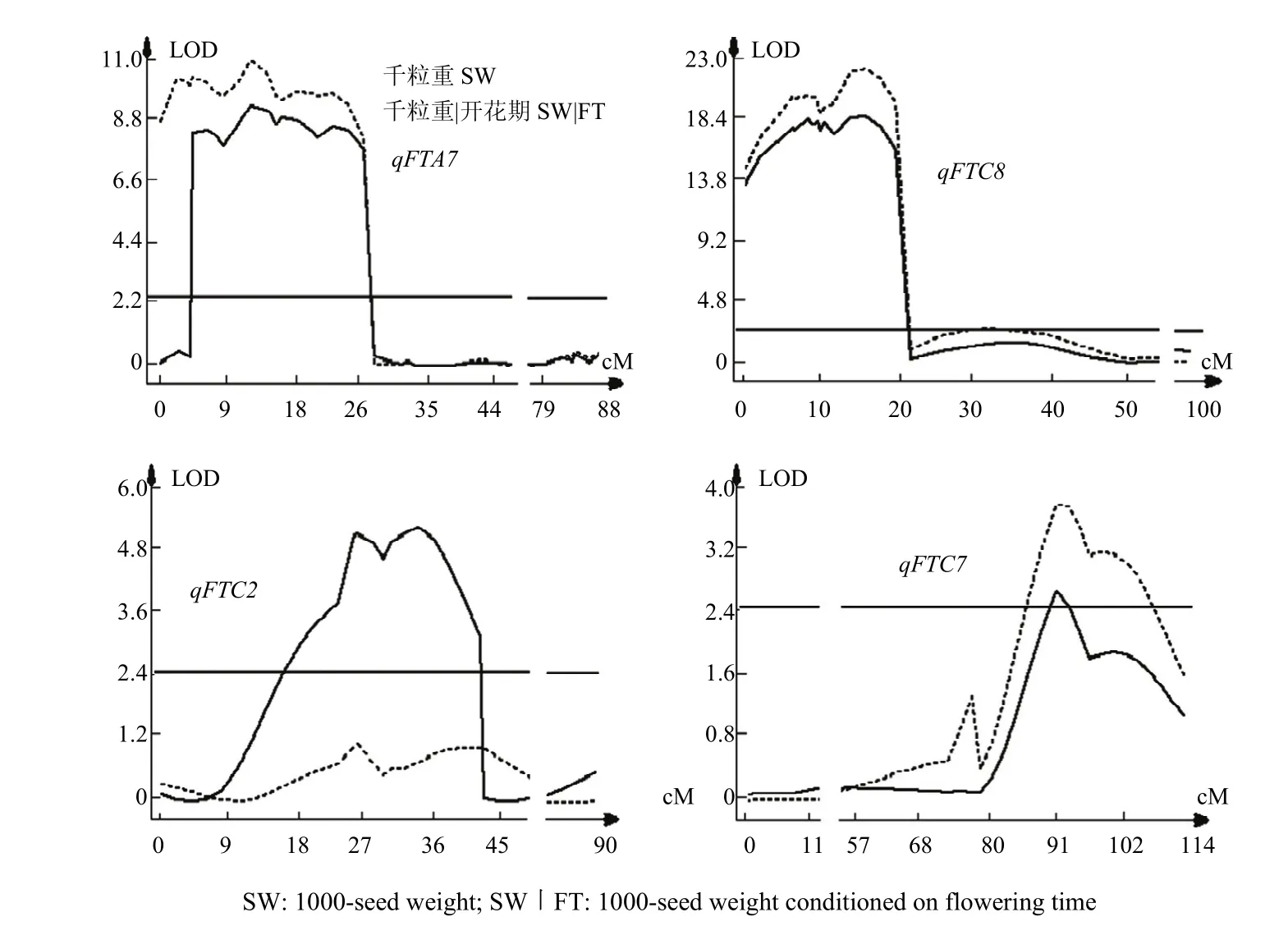
图2 开花期对SG群体中4个主效千粒重QTL的影响模式Fig. 2 The genetic regulation of flowering time to 1000-seed weight on four major QTL for seed weight in SG population
2.5 主效开花期QTL连锁标记的育种应用
为验证主效开花期QTL的遗传效应,探讨通过标记筛选,聚合提早开花等位基因,获得早熟育种材料的可能性。根据SG群体282个株系在9种环境下的开花期平均值,选取开花最早和最迟的株系各20个,用效应值最大、显著性最高的3个主效QTL(qFTA2、qFTC2和qFTC6)峰值及相近位置12个标记进行基因型和性状表现型的关联分析,从中筛选出6个高度关联的共显性标记(表5)。在播种到开花平均为176 d的 20个最早开花的株系中,qFTA2连锁标记ZAAS548b和DNAPL的基因型70%和80%显示早开花等位基因带型,qFTC2和qFTC6各2个标记符合率则达到80%和95%;在播种到开花平均为192 d的20个最迟开花的株系中,qFTA2位点2标记显示80%和75%条带为迟开花基因型而qFTC2和qFTC6位点,迟开基因型高达100%和90%。因此,选用这些标记转育早花早熟基因尤其是通过回交和复交,将中国油菜中的早熟基因导入和聚合于欧洲油菜品种中,创制适合长江流域油菜主产区种植环境而以欧洲油菜为主要遗传背景的育种新资源,在杂交油菜育种中将发挥重要作用。另最早和最迟开花的各20个株系,平均千粒重差值达0.328 g,差异极显著(P=0.015);但早花和迟花二组间含油量和角果粒数均无显著差异(P=0.274和0.189)。
3 讨论
3.1 不同遗传背景下开花期QTL比较

表4 千粒重条件和非条件QTL 的加性效应Table 4 Additive effect of conditional and unconditional QTLs for 1000-seed weight
本研究在ZHAO等[2]前期研究基础上,利用新增6种环境的开花期表型数据和添加356个分子标记后的新版SG图谱,对共9种环境下的开花期性状重新进行QTL扫描,共检测到在3种以上环境中稳定表达的QTL有7个。其中,3个效应值最大的QTL是qFTA2、 qFTC2和qFTC6,Gaoyou等位基因纯合并存时,可相应提早开花约3、6和4 d,是创制油菜早熟亲本的候选QTL位点。油菜开花期QTL的研究已有众多报道,几乎在所有 19条连锁群上均检测到相关基因位点,但出现频率最高、效应值最大的QTL主要分布在A2、C2和C6连锁群上[1,5-9],与本研究结果相吻合。比较先前3环境下的定位结果[1],qFTA2、qFTC2和qFTC6的显著性和效应值均有提高,qFTA3、qFTA4、qFTA10和qFTC8是新定位的QTL,分别在6、4、5 和3种环境中表达显著,而之前在3种环境联合定位分析中检测到的A1、C1、C4和C9上的QTL只在1 —2种环境中显著,未计入在内。这可能是随着图谱上标记饱和度的增加和全基因组覆盖率的提高,以及试验环境数的增加和试验误差减小,使得统计精确性加强,QTL的检出率提高而只在少数环境下检测到的效应值又较小的QTL很可能是假阳性QTL。
包括油菜在内的多个芸薹属作物全基因组的测序完成为QTL的精细定位、候选基因筛选和图位克隆提供了强大的信息资源和技术支撑。通过序列比对,首先将qFTA2、qFTC2和qFTC6确定在油菜或甘蓝基因组相应物理区间,分别位于油菜ChrA2∶4—8 Mb、甘蓝C02∶1—4.5 Mb和油菜ChrC6∶25—29 Mb区间。拟南芥基因组同源比对,在qFTA2和qFTC6峰值位置发现拟南芥中调控开花关键基因FLOWERING LOCUS T(FT)的2个同源栲贝BnFT-A2(GSBRNA2T00090951001)和BnFT-C6(GSBRNA2T00067517001)。BnFT-C6两侧还发现BnAP1-1(GSBRNA2T00118005001)和BnAP1-2 (GSBRNA2T00054833001)2个开花相关基因,这个结果与LONG等[9]和WANG等[14]的报道相一致(图1-a和图1-c)。除此,拟南芥中“春化途径”的重要基因 FLOWERING LOCUS C(FLC)的同源基因BnFLC-C2 (GSBRNA2T00068991001)和与开花有关的酵母多聚腺苷酸化因子 pfs2p同源基因 BnFY-C2 (GSBRNA2T00151903001)位于 qFTC2区间峰值位置(图1-b),该区间同源于KOORNNEEF等[32]早年报道的拟南芥第5染色体上包含FLC、CO和FY等开花基因的区域,与 RAMAN等[17]报道的贡献率最大(22.39%)的QTL Qdtf(g).wwai-C2a,可能属于相同QTL。研究结果为后续功能确认提供重要参考。
3.2 开花期QTL对环境的响应以及基因间的互作
9环境下检出的在3种以上环境中表达的7个开花期QTL,除qFTA10中国西安环境中被检测到微效QE互作,其余QTL与环境的互作效应均未达显著,上位性QTL与环境互作也只在德国Reinshof环境下,ZAAS1029/qFTC2位点检测到微效的AAE互作,说明开花期是一个相对稳定的性状,环境互作不大。8对互作上位性QTL,效应均较微,累计不到加性主效的一半,值得注意的是7个加性主效QTL中,除了qFTC8外,其余 6个均有上位性效应,2个最重要的QTLqFTC2和qFTC6分别与4和2个QTL/标记位点发生互作,qFTA2、qFTA3和qFTA10也分别参与上位互作1—2次。因此,借助标记辅助转育早熟基因时因考虑到与其他QTL或位点的互作而带来的影响。
开花性状在模式植物拟南芥中已研究得相当清楚,由几百个基因共同参与,是一个复杂的基因网络[16],如“春化途径”关键基因 FLC同时又受到SHORTVEGETATIVEPHASE(SVP)、TEMPRANILLO1 (TEM1)、SUPPRESSOR、OFFRIGIDA4(SUF4)、FRIGIDA(FRI)、EARLYFLOWERING7(ELF7)、EARLYFLOWERINGINSHORTDAYS(EFS)、VERNALIZATION INSENSITIVE 3(VIN3)、VERNALIZATION2(VRN2)等多个基因、多条途径调控,因而含BnFLC-C2(GSBRNA2T00068991001)的qFTC2区域参与多个上位互作是可以理解的。
3.3 开花期对种子千粒重的影响
比较千粒重在非条件和给定开花期为条件时的QTL分析结果,可以看出开花期对千粒重QTL的作用模式有 3种情况。(1)当开花期被条件后,LOD值急剧下降至阀值以下,QTL效应不再显著,如效应值较小的4个QTL qSWA2、qSWA3、qSWA4和qSWC2,大粒等位基因均来自中国亲本Gaoyou。其中qSWC2 与qFTC2同位点,qSWA2和qSWA3相邻处有qFTA2 和qFTA3 2个开花期QTL,早开花等位基因均来自Gaoyou,显然在这些位点上,大粒等位基因效应可能与开花早、籽粒灌浆期长有关。当早开花优势消失后,粒重QTL不覆存在。通过选择这些位点的早开花标记基因型有望同时提高种子千粒重,这也部分给出了开花期与千粒重之间极显著负相关的遗传解释;(2)开花期的变化对粒重 QTL基本无影响,如 qSWA1、qSWA7、qSWC7和qSWC8,LOD值虽出现小幅上升,效应值基本不变。除qSWC8,其他3个QTL位点,欧洲亲本Sollux增加粒重。qSWA7和qSWC8是SG群体中2个效应值最大,显著性最高的QTL,当给定开花期不变时,LOD值和贡献率有所提高,效应值稳定。因此,这两个效应值最大的粒重QTL推荐育种应用可有效提高千粒重但不影响开花期;(3)如qSWC1-1和qSWC1-2,当开花期给定不变时,QTL的LOD值下降或效应值同时下降,说明开花迟早对其在一定范围内有一定影响。
3.4 QTL连锁标记的筛选和育种潜力分析
根据SG群体极端开花期株系在3个效应值最大的QTL(qFTA2、qFTC2和qFTC6)区域标记基因型和开花期表现型的关联分析(表5),筛选获得6个高质量、高吻合度的共显性标记。基于这3个QTL的标记辅助,目前,笔者正在试图通过回交导入和自交纯合,将中国油菜中的早花早熟基因导入和聚合到欧洲油菜遗传背景中,创制出以优良欧洲油菜品种为遗传背景但开花期可提早约半个月的育种新材料,这对扩大中国油菜亲本材料的遗传基础,增加强优势组合的父本来源产生积极作用,是分子育种设计和创新育种理念的新途径和新思路。这些标记目前已投入育种应用并收到了较好的选育效果(数据未列出)。
另外。本研究发现迟、早开花组各20个株系除开花期相差19 d,早开花组千粒重平均值高于迟开花组0.3218 g,差异极显著,但含油量和角果粒数基本持平。因此,通过标记辅助导入早花基因可同时提高千粒重,但不影响含油量和角果粒数,对群体产量的正向作用有待下一步试验确认。
4 结论
7个QTL均显示早开花等位基因来自中国亲本。拟南芥中调控开花关键基因FT、API、FLC和FY的6个同源拷贝定位到了3个主效QTL峰值位置。开花迟、早显著影响4个千粒重QTL,但2个最重要的粒重位点(qSWA7和 qSWC8)不受影响。3个主效 QTL (qFTA2、qFTC2和qFTC6)的6个共显性标记可用于早熟基因的导入和早熟材料的选育。
References
[1] ZHAO J Y, BECKER H C, DING H D, ZHANG Y F, ZHANG D Q,ECKE W. QTL of three agronomically important traits and their interactions with environment in a European×Chinese rapeseeed population. Acta Genetica Sinica, 2005, 32(9): 969-978.
[2] 刘后利. 几种芸薹属油菜的起源和进化. 作物学报, 1984, 10(1):9-18. LIU H L. Origin and evolution of Brassicarape. Acta Agronomica Sinica, 1984, 10(1): 9-18. (in Chinese)
[3] DETJEN L R. A preliminary report on cabbage breeding. Proceedings of the American Society for Horticultural Science, 1926, 23: 325-332.
[4] DICKSON M H. Eight newly described genes in broccoli. Proceedings of the American Society for Horticultural Science, 1968,93: 356.
[5] TEUTONICO R A, OSBORN T C. Mapping loci controlling vernalization requirement in Brassica rapa. Theoretical and Applied Genetics, 1995, 91(8): 1279-1283.
[6] FERREIRA M E, SATAGOPAN J, YANDELL B S, WILLIAMS P H,OSBORN T C. Mapping loci controlling vernalization requirement and flowering time in Brassica napus. Theoretical and Applied Genetics, 1995, 90(5): 727-732.
[7] OSBORN T C, KOLE C, PARKIN A P, SHARPE A G, KUIPER M,LYDIATE D J, TRICK M. Comparison of flowering time genes in Brassica rapa, B. napus and Arabidopsis thaliana. Genetics, 1997,146(3): 1123-1129.
[8] A Linkage analysis of molecular markers and quantitative trait loci in populations of inbred backcross lines of Brassica napus L.. Genetics,1999, 153(2): 949-964.
[9] LONG Y, SHI J, QIU D, LI R, ZHANG C, WANG J, CHOI S R. Flowering time quantitative trait loci analysis of oilseed Brassica in multiple environments and genome wide alignment with Arabidopsis. Genetics, 2007,177(4): 2433-2444.
[10] 蔡长春, 傅廷栋, 陈宝元, 涂金星. 甘蓝型油菜遗传图谱的构建及开花期的QTL分析. 中国油料作物学报, 2007, 29(1): 1-8. CAI C C, FU T D, CHEN B Y, TU J X. Construction of a genetic linkagemap and its use for QTL analysis of flowering time in Brassica napus L.. Chinese Journal of Oil Crop Science, 2007, 29(1): 1-8. (in Chinese)
[11] XU L P, HU K N, ZHANG Z Q, GUAN C Y, CHEN S, HUA W, LI J N, WEN J, YI B, SHEN J X , MA C Z, TU J X , FU T D . Genome-wide association study reveals the genetic architecture of flowering time in rapeseed (Brassica napus L.). DNA Research, 2016,23(1): 43-52.
[12] WEI D, MEI J, FU Y, DISI J O, LI J, QIAN W. Quantitative trait loci analyses for resistance to Sclerotinia sclerotiorum and flowering time in Brassica napus. Molecular Breeding, 2014, 34(4): 1797-1804.
[13] NELSON M N, RAJASEKARAN R, SMITH A, CHEN S, BEECK C P, SIDDIQUE K H, COWLING W A. Quantitative trait loci for thermal time to flowering and photoperiod responsiveness discovered in summer annual-type Brassica napus L.. PLoS One, 2014, 9(7):e102611.
[14] WANG J, LONG, Y, WU B, LIU J, JIANG C, SHI L, ZHAO J ,GRAHAM J K, MENG J. The evolution of Brassica napus FLOWERING LOCUS paralogues in the context of invertedchromosomal duplication blocks. BMC Evolutionary Biology, 2009,9(1): 271.
[15] ZOU J, RAMAN H, GUO S, HU D, WEI Z, LUO Z, SHI W, FU Z,DU D, MENG J. Constructing a dense genetic linkage map and mapping QTL for the traits of flower development in Brassica carinata. Theoretical and Applied Genetics, 2014, 127(7): 1593-1605.
[16] FORNARA F, MONTAIGU A, COUPLAND G. SnapShot: Control of flowering in Arabidopsis. Cell, 2010, 141(3): 550-550.
[17] RAMAN H, RAMAN R, ECKERMANN P, COOMBES N, MANOLI S, ZOU X, BATLEY J. Genetic and physical mapping of flowering time loci in canola (Brassica napus L.). Theoretical and Applied Genetics, 2013, 126(1): 119-132.
[18] HOU J, LONG Y, RAMAN H, ZOU X, WANG J, DAI S, XIAO Q ,LI C, FAN L , LIU B,MENG J. A Tourist-like MITE insertion in the upstream region of the BnFLC. A10 gene is associated with vernalization requirement in rapeseed (Brassica napus L.). BMC Plant Biology, 2012, 12: 238.
[19] 刘玉霞, 汪义龙, 丁瑜, 陈飞, 黄吉祥, 倪西源, 赵坚义. 油菜种子成熟度对千粒重和含油量性状的影响. 浙江农业学报, 2011, 23(3):465-469. LIU Y X, WANG Y L, DING Y, CHEN F, HUANG J X, NI X Y,ZHAO J Y. Effects of seed maturity on seed weight and oil content in Brassica napus. Acta Agiculturae Zhejiang Gensis, 2011, 23(3):465-469. (in Chinese)
[20] 郑本川, 张锦芳, 李浩杰, 蒲晓斌, 崔成, 柴靓, 蒋俊, 牛应泽, 蒋梁材. 甘蓝型油菜生育期天数与产量构成性状的相关分析. 中国油料作物学报, 2013, 35(3): 240-245. ZHENG B C, ZHANG J F, LI H J, PU X B, CUI C, CHAI L, JIANG J,NIU Y Z, JIANG L C. Correlation between duration of growth periods and yield components of Brassica napus L.. Chinese Journal of Oil Crop Sciences, 2013, 35(3): 240-245. (in Chinese)
[21] ZHANG L W, YANG G S, LIU P W, HONG D F, LI S P, HE Q B. Genetic and correlation analysis of silique-traits in Brassica napus L. by quantitative trait locus mapping. Theoretical and Applied Genetics,2011, 122(1): 21-31.
[22] YANG P, SHU C, CHEN L, XU J S, WU J, LIU K D. Identification of a major QTL for silique length and seed weight in oilseed rape (Brassica napus L.). Theoretical and Applied Genetics, 2012, 125(2):285-296.
[23] FAN C, CAI G, QIN, J, LI Q, YANG M, WU J, FU T, LIU K, ZHOU Y. Mapping of quantitative trait loci and development of allele-specific markers for seed weight in Brassica napus. Theoretical and Applied Genetics, 2010, 121(7): 1289-1301.
[24] LI N, SHI J, WANG X, LIU G, WANG H. A combined linkage and regional association mapping validation and fine mapping of two major pleiotropic QTLs for seed weight and silique length in rapeseed (Brassica napus L.). BMC Pant Biology, 2014, 14(1): 1-14.
[25] LIU J, HUA W, HU Z, YANG H, ZHANG L, LI R, DENG L , SUN X,WANG X, WANG H. Natural variation in ARF18 gene simultaneously affects seed weight and silique length in polyploid rapeseed. Proceedings of the National Academy of Sciences of the USA, 2015, 112(37): E5123-E5132.
[26] ZHAO J Y, HUANG J X, CHEN F, XU F, NI X, XU H, WANG Y,JIANG C, WANG H, XU A, HUANG R, LI D, MENG J. Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theoretical and Applied Genetics, 2012, 124(2):407-421.
[27] ZHU J. Analysis of conditional genetic effects and variance components in developmental genetics. Genetics, 1995, 141(4):1633-1639.
[28] WANG S C, BASTERN J, ZENG Z B. Window QTL Cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University, 2007, http://Statgen.ncsu.edu/ qtlcart/ WQTLCart. htm.
[29] YANG J, ZHU J, WILLIAMS R W. Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics, 2007,23(12): 1527-1536.
[30] WANG D L, ZHU J, LI K L, PATERSON A H. Mapping QTLs with epistatic effects and QTL× environmrnt interactions by mixed linear model approaches. Theoretical and Applied Genetics, 1999, 99(7/8):1255-1264.
[31] CHALHOUB B, DENOEUD F, LIU S, PARKIN I A, TANG H,WANG X, CORRÉA M. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science, 2014, 345(6199):950-953.
[32] KOORNNEEF M, VRIES H, HANHART C, SOPPE W, PEETERS T. The phenotype of some late-flowering mutants is enhanced by a locus on chromosome 5 that is not effective in the Landsberg erecta wild-type. The Plant Journal, 1994, 6(6): 911-919.
(责任编辑 李莉)
Mapping QTL of Flowering Time and Their Genetic Relationships with Seed Weight in Brassica napus
HUANG Ji-xiang1, XIONG Hua-xin1,2, PAN Bing1,3, NI Xi-yuan1, ZHANG Xiao-yu1, ZHAO Jian-yi1
(1Institute of Crop and Nuclear Technology Utilization, Zhejiang Academy of Agricultural Sciences/State Key Laboratory Breeding Base for Zhejiang Sustainable Pest and Disease Control, Hangzhou 310021;2College of Chemistry and Life Sciences, Zhejiang Normal University, Jinhua 321000, Zhejiang;3College of Agriculture & Biotechnology, Zhejiang University, Hangzhou 310058)
【Objective】The present research aimed to explore the major QTL controlling the flowering time in European andChinese rapeseed materials, to analyze the genetic influence of flowering time on QTL for 1000-seeds weight, and thus to provide available information for breeding purpose.【Method】The doubled haploid (DH) Sollux/Gaoyou population with 282 lines was used. The data set of flowering time was obtained from nine environments and over seven years. QTL identification of flowering time was performed using WinQTLCart 2.5. The candidate genes underlining QTLs were screened out by transcriptome analysis using RNA-Seq and alignment between QTL regions and Arabidopsis. Further, conditional QTL estimation was adopted to dissect the genetic relationships between flowering time and seed weight. Finally, using selected DH lines with extreme phenotypes of flowering time, an association evaluation between marker genotypes and phenotypes of flowering time was performed. 【Result】 Seven major QTLs were detected, which showing significant at least in three environments. Their additive effects ranged from 0.58-3.85 days and together accounted for around 84% of the phenotypic variation in population. The sum of eight pairs of epistatic loci (additive × additive) accounted for 41.8% of the total additive effects. QTL by environmental interactions were significant only in few environments with small amount of genetic effects. Four critical orthologous genes of Arabidopsis thaliana for flowering time were mapped in the peak positions of three most significant QTL regions (qFTA2, qFTC2, and qFTC6). It provides valuable information to anchor candidate genes underling QTL. The conditional QTL analysis revealed large impact of flowering time on seed weight in four QTLs (qSWA2, qSWA3, qSWA4, and qSWC2). This partly explained the significant negative correlation between flowering time and 1000-seed weight. While the most important two (qSWA7 and qSWC8) showed independent without being interfered. Six markers linked with three major QTLs showed good fitness between marker genotypes and trait phenotypes (70%-100%), indicating their potentials for breeding purpose. The results demonstrated that the combination of early flowering alleles from qFTA2, qFTC2 and qFTC6 by marker assistant selection of ZAAS548, DNAPL, ZAAS619sa, ZAAS616s, ZAAS846a and C6SGFLO-22 induced not only early flowering but also significantly increased 1000-seed weight, while the oil content and seeds per silique between two extreme flowering time groups showed almost the same.【Conclusion】All seven QTLs of flowering time showed Chinese parent Gaoyou induced early flowering. Four important candidate genes homologous to Arabidopsis controlling flowering time (FT, FLC,AP1, and FY) were physically aligned and mapped underlining the peak positions of the three major QTL qFTA2, qFTC2 and qFTC6. The results indicated that the four loci corresponding to seed weight were genetically influenced by flowering time, however, the most important two (qSWA7 and qSWC8) were independent. Six markers linked to the 3 major QTL were of potentials in the practical breeding program.
Brassica napus L.; QTL mapping; flowering time; 1000-seeds weight; conditional QTL mapping
2016-02-25;接受日期:2016-05-10
国家重点基础研究发展计划(973计划)(2015CB150200)、七大农作物育种专项(JFYS2016ZY03002156)、浙江省旱粮创新团队项目(2011R50026-7)
联系方式:黄吉祥,Tel:0571-86403406;E-mail:838107@163.com。熊化鑫,Tel:0571-86403406;E-mail:huaxinxiong_11@163.com。黄吉祥和熊化鑫为同等贡献作者。通信作者赵坚义,Tel:0571-86403406;E-mail:2208086097@qq.com
