致阴道病的白念珠菌多位点序列分型分析及遗传多样性
2016-08-08王志恒应春妹
王志恒,应春妹,赵 虎
致阴道病的白念珠菌多位点序列分型分析及遗传多样性
王志恒1,应春妹2,赵虎1
摘要:目的 对上海市3所妇产科医院分离的114株白念珠菌进行分子流行病学研究,分析本地专科医院分离的白念珠菌主要基因型别与菌株耐药关系,并了解菌株间遗传多样性与种群分类关系。方法 收集来自复旦大学附属妇产科医院、上海市第一妇幼保健院和国际和平妇幼保健院共114株白念珠菌,采用eBURST进行菌株亲缘性分析,多位点序列分型(MLST)方法进行分型,并采用ATBTM FUNGUS 3试剂盒作真菌体外药物敏感试验。结果 114株菌株中共有47种DST型(diploid strain types, DST),其中已知30种,主要组群为DST 79和DST 435。114株白念珠菌对氟胞嘧啶、两性霉素B、氟康唑、伊曲康唑、伏立康唑敏感率分别为96.5%、100%、85.1%、55.2%、84.3%。结论 上海地区不同妇产科医院致病性白念珠菌呈多克隆系,但主要为DST 79和DST 435型,且具有一定耐药性。通过应用MLST分型方法,初步发现上海地区白念珠菌具有遗传多样性,且种群分类与基因分型具有相关性。
关键词:白念珠菌; 多位点序列分型; 基因分型
念珠菌性阴道病是一种常见妇科疾病,发病率仅次于细菌性阴道病。有研究表明,念珠菌性阴道病致病念珠菌85%~95%为白念珠菌[1]。白念珠菌阴道病危害严重,不仅会造成阴道瘙痒引起生活质量下降,而且可以通过影响精子运动引发不孕,极少数患者会导致早产及胎儿发育不良,也可引起宫颈糜烂、输卵管炎及盆腔炎[2]。随着临床抗真菌药物的广泛应用,真菌耐药性也越益严重。对真菌耐药基因的检测有助于抗真菌药物的选择,但真菌基因型随着环境与时间的变化会产生一系列突变,最终导致其耐药性的产生[3]。多位点序列分型(MLST)是一种基于核酸序列测定的分型方法,能对基因结构高度多态性的致病性真菌进行分型和遗传分组,并能很好地区分来自同一种群或地区的菌株,其具有很高的分辨能力,适于分子流行病学研究和分子进化学研究。本研究采用MLST方法对3所医院114株引起念珠菌阴道病的白念珠菌进行基因分型并探索其基因的变化过程。
1 材料与方法
1.1菌株来源及鉴定保存
收集2013年11月—2014年1月临床分离白念珠菌114株,其中 29株来自国际和平妇婴保健院,26株来自上海市第一妇婴保健院,59株来自复旦大学附属妇产科医院,菌株均分离自患有念珠菌性阴道病女性的阴道分泌物,患者年龄21~56岁。阴道分泌物标本收集后立即接种于 YPD(yeast extract peptone dextrose medium,YPD,上海生工生物工程科技有限公司)琼脂平皿和科玛嘉显色平皿(上海科玛嘉微生物技术有限公司),置于35℃、5% CO2孵育箱中培养24 ~48 h,YPD琼脂平皿上菌落呈奶酪色,表面湿润光滑且边缘整齐;科玛嘉显色平皿上菌落呈翠绿色;涂片镜检,见镜下圆形或卵圆形菌体或孢子及假菌丝,则鉴定为白念珠菌。
1.2方法
1.2.1 DNA制备、扩增和测序 使用Biospin真菌基因组DNA抽提试剂盒(BioFlux公司)抽提白念珠菌基因组DNA, NanoDrop2000测量DNA浓度,用双蒸水稀释至100~200 ng/μL。本实验中使用白念珠菌的7个管家基因及其PCR扩增引物见表1。PCR总反应体系为30 μL,包括:基因组DNA 50 ng,2×MasteMix 15 μL, 10 μmol/L上下游引物各0.5 μL,用去离子水补充至30 μL。PCR 反应条件为:95℃ 5 min,95℃ 30 s,60℃30 s,72℃ 50 s,35个循环。PCR产物测序由华大基因公司完成。1.2.2 测序结果分析和MLST 利用BioEdit以及DNAStar对菌株管家基因的测序结果进行阅读和比对;将7个管家基因与数据库上相应的等位基因进行比较,分别获得每个菌株的等位基因型,并确定其DST型(diploid strain types, DST)。

表1 白念珠菌7种管家基因引物序列Table 1 Primers of 7 housekeeping genes of Candida albicans
1.2.3 菌株进化关系分析 采用eBURST进行菌株亲缘性分析,并参照网上数据库中下载的所有白念珠菌MLST信息,利用SPSS17.0统计软件对菌株进行聚类分析和数据分析。
1.2.4 真菌体外药物敏感试验 菌株分离纯化后,使用酵母样真菌敏感性检测试剂盒(ATBTMFUNGUS 3,法国生物梅里埃公司)鉴定。该试剂盒包括氟胞嘧啶、两性霉素B、氟康唑、伊曲康唑和伏立康唑5种抗真菌药物。以近平滑念珠菌ATCC 22019和克柔念珠菌ATCC 6258作为质控菌株。
2 结果
2.1MLST分型结果
通过对114株白念球菌的7个管家基因的测序结果分析和在线比对,97株菌株为网站数据库中已登记DST型,见表2。17株可能为新DST型,网站数据库中尚未登记。
2.2菌株亲缘性分析
由于17株菌落可能的DST型未在数据库中登记,其DST型还没有确定,因此,只对已登记的97株菌株进行分析。97株DST型菌株中,有30种不同的基因型。通过eBURST分析,30种基因型的白念球菌分2个主要克隆群:DST 79和DST 435,其中DST 79共有40株,占总数的35.1%(40/114),DST 435有 14株, 占 12.3% (14/114)。DST 1395有7株,DST 2627有4株,其他DST型菌株单个或双个分散存在,为独特型,见图1。
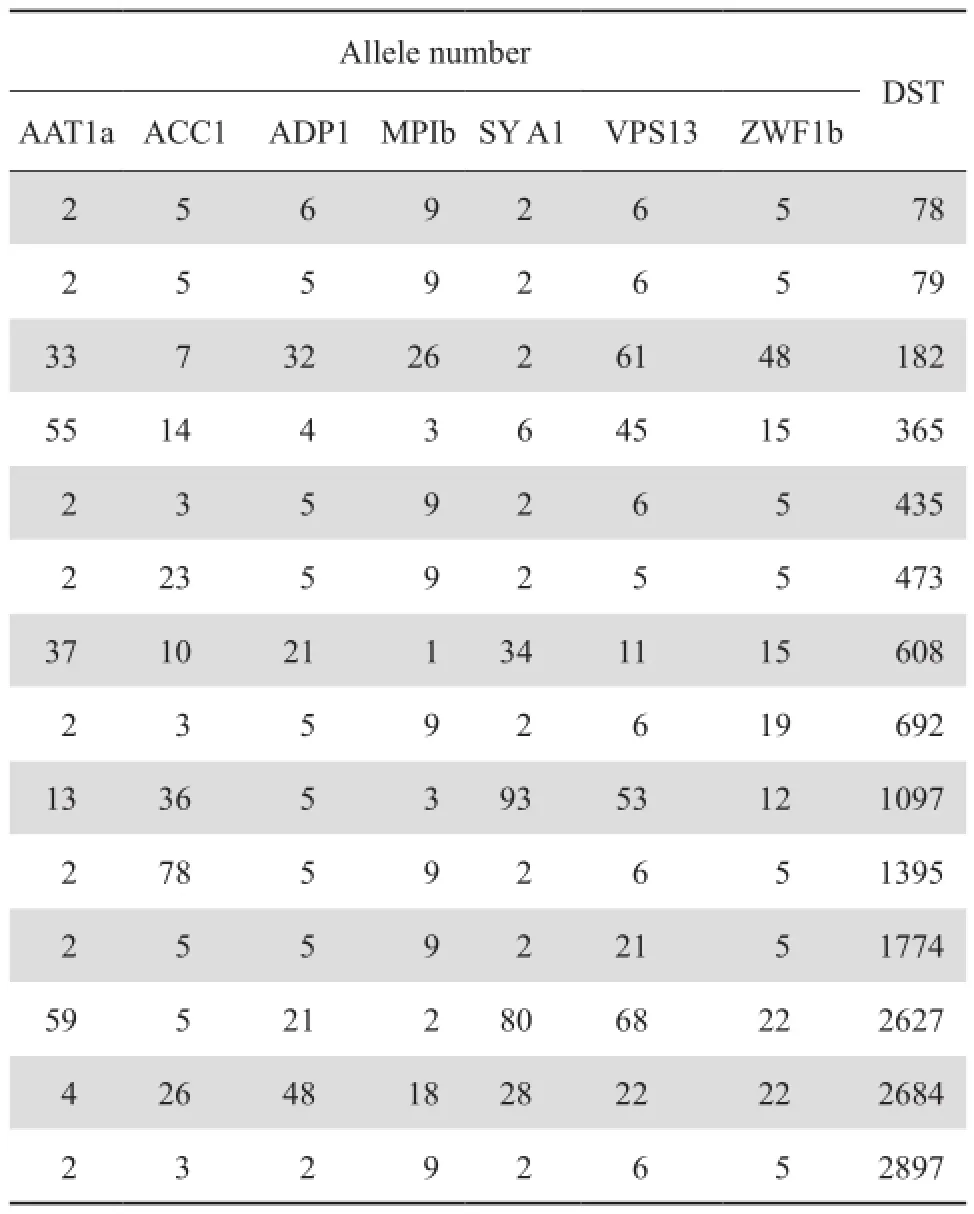
表2 部分白念珠菌等位基因数和DST型Table 2 Allele number of selected genes and DST type of Candida albicans
通过与数据库(http://pubmlst.org/calbicans/)中世界各地不同DST型菌株进行eBURST比对分析发现,菌株群可分为多个分支,主要克隆群是DST 69,由DST 69菌株群向其他类型菌株分离进化。我们实验中得到的DST 79是DST 69的一个直接分支,再由DST 79向DST 435方向进一步分型,见图2。
2.3体外药物敏感性试验
114株白念珠菌中,所有菌株对两性霉素B敏感,敏感率100%;110株对氟胞嘧啶敏感,占96.5%;97株对氟康唑敏感,占85.1%;63株对伊曲康唑敏感,占55.2%;96株对伏立康唑敏感,占84.3%。见表3。
2.4菌株进化与耐药性关系分析
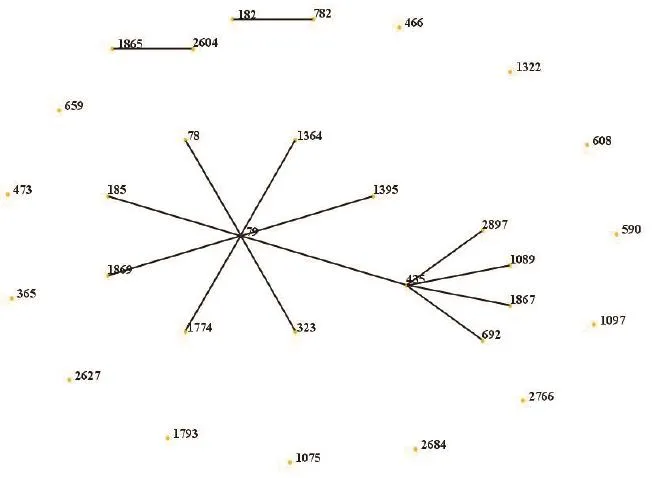
图1 30种DST型菌株eBURST亲缘性分析Figure 1 Phylogenetic analysis of 30 DST isolates by eBURST
利用SPSS对114株白念球菌进行聚类分析,采用Between-groups linkage聚类方法分析,结果见图3。所有菌株主要分为8个分支,其中,主要克隆群DST 79和DST 435处于同一组中,其余分散存在的克隆以及未知DST型克隆分别在其他7个分支中。

图2 DST 69、DST 79和DST 435克隆群分布图Figure 2 Colony distribution of DST 69, DST 79 and DST 435

表3 114株白念珠菌对5种抗真菌药物敏感率Table 3 Susceptibility of 114 strains of Candida albicans to 5 antifungal agents
将DST聚类分析与其相应的耐药性相比较,主要的耐药菌株类型为DST 79、DST 435和DST 608等,见表4。通过将3所医院取得的菌株分别统计后发现,复旦大学附属妇产科医院分离的菌株DST型种类最多,且主要的耐药菌株类型是DST 79和DST 435;第一妇婴保健院主要的耐药菌株类型是DST 79;国际和平妇婴保健院耐药株比例较低,主要也为DST 79株。
3 讨论
MLST作为基因分型的标准方法被广泛应用于微生物感染的分子流行病学研究中[4]。本研究采用MLST方法分析了上海3所妇产科医院阴道分离的114株白念珠菌,主要结果:①分离自患者阴道分泌物的致病白念珠菌主要类型是DST 79 和DST 435克隆群;②DST 79和DST 435克隆群属于祖源型菌株DST 69的一个分支型,也是本实验中主要耐药型菌株;③菌株主要分布于8个分支,DST 79和DST 435集中在同一分支,其余分散存在的克隆以及未知DST型克隆分别分在另外7个分支中。
本实验发现,耐药菌株的基因型集中在DST 79、DST 435、DST 608、DST 1395、DST 2627 和DST 2897基因型,但这6种基因型菌株耐药率并不是100%,且对不同药物的耐药率不同。DST 608型菌株虽然对其中3种药物的耐药率为100%,但由于该基因型菌株只有1株,不足以说明该基因型的确切耐药情况。TAVANTI等[5]和ODDS 等[6]利用MLST方法也有类似的状况,耐药基因型也是散在不同分支中,且同一分支菌株耐药率并不是100%。导致同类型菌株不同耐药性的原因可能是不同基因型如特定位点杂合子或纯合子差异等。

图3 MLST分型结果聚类分析图Figure 3 Cluster analysis of genotyping results of multilocus sequence typing

表4 不同DST型耐药菌株统计Table 4 Summary of drug resistant strains in terms of DSTs
祖源型菌株DST 69菌株主要分离自美国、日本、马来西亚、中东、新西兰、英国和欧洲等[7- 8]国家和地区,而中国的白念珠菌主要以其分支为主,很少发现DST 69菌株。经过比对,WU等[9]在上海某医院中分离的白念珠菌DST 型在本实验所有的分支中都有,表明上海地区人群中的白念珠菌具有较强的一致性。GONG等[10]通过对取自沈阳人群消化道的白念珠菌进行MLST发现,DST 1593和DST 142等是最常见的基因型,由此可见,不同地区的致病白念珠菌基因型群具有较大的差异。SHIN等[11]发现,从韩国人群中分离的菌株主要类型为DST 69和DST 732,包括了来自中国、韩国、日本等亚洲国家的菌种以及西方的主要菌种,表明白念珠菌可能随着人群流动而播散。
白念珠菌作为一种条件致病性真菌,其基因组具有很大的变化潜能,经过环境选择的压力,其染色体更易产生重组从而进化出具有较强的致病性特征[12]。因此,对白念珠菌的种群结构、流行病学调查和念珠菌属物种的系统发育分析,为临床提供了有用的诊断信息。本实验中发现的致病菌基因种类多样,耐药菌株种类集中,且主要为对伊曲康唑耐药,而对氟胞嘧啶和两性霉素B耐药性非常低,因此,检测白念珠菌类型可以更好地指导临床用药。
参考文献:
[1]SOBEL JD. Vulvovaginal candidosis [J]. Lancet, 2007,369(9577): 1961-1971.
[2] TIAN YH, XIONG JW, HU L, et al.Candida albicans and fltrates interfere with human spermatozoal motility and alter the ultrastructure of spermatozoa: an in vitro study [J]. Int J Androl,2007, 30(5): 421-429.
[3]GUZEL AB, DOGEN A, AYDIN M, et al. Genotyping reveals no link between Candida albicans genotype and vaginitis severity in Turkish women [J]. Mycopathologia, 2013, 175(3-4): 287-294.
[4]Cliff PR, SANDOE JA, HERITAGE J, et al. Use of multilocus sequence typing for the investigation of colonisation by Candida albicans in intensive care unit patients [J]. J Hosp Infect, 2008,69(1): 24-32.
[5]TAVANTI A, DAVIDSON AD, FORDYCE MJ, et al. Population structure and properties of Candida albicans, as determined by multilocus sequence typing [J]. J Clin Microbiol, 2005, 43(11):5601-5613.
[6]ODDS FC, BOUGNOUX ME, SHAW DJ, et al. Molecular phylogenetics of Candida albicans [J]. Eukaryot Cell, 2007, 6(6):1041-1052.
[7]ODDS FC. Molecular phylogenetics and epidemiology of Candida albicans [J]. Future Microbiol, 2010, 5(1): 67-79.
[8]TAVANTI A, DAVIDSON AD, FORDYCE MJ, et al. Population structure and properties of Candida albicans, as determined by multilocus sequence typing [J]. J Clin Microbiol, 2005, 43(11):5601-5613.
[9]WU K, LUO T, LI L, et al. Multilocus sequence typing of pathogenic Candida albicans isolates collected from a teaching hospital in Shanghai, China: A molecular epidemiology study [J]. PLoS One, 2015, 10(4): e125245.
[10] GONG YB, ZHENG JL, JIN B, et al. ParticularCandida albicans strains in the digestive tract of dyspeptic patients, identifed by multilocus sequence typing [J]. PLoS One, 2012, 7(4): e35311.
[11]SHIN JH, BOUGNOUX ME, D'ENFERT C, et al. Genetic diversity among Korean Candida albicans bloodstream isolates:assessment by multilocus sequence typing and restriction endonuclease analysis of genomic DNA by use of BssHII [J]. J Clin Microbiol, 2011, 49(7): 2572-2577.
[12]MCMANUS BA, COLEMAN DC. Molecular epidemiology,phylogeny and evolution of Candida albicans [J]. Infect Genet Evol, 2014, 21: 166-178.
2.复旦大学附属妇产科医院检验科。
中图分类号:R379.4
文献标识码:A
文章编号:1009-7708(2016)03-0330-06
DOI:10.16718/j.1009-7708.2016.03.014
收稿日期:2015-08-31 修回日期:2015-12-02
作者单位:1.复旦大学附属华东医院检验科,上海 200040;
作者简介:王志恒(1986—),男,硕士,初级技师,主要从事细菌耐药性监测和耐药机制研究。
通信作者:赵虎,E-mail:hubertzhao@163.com。
Multilocus sequence typing and genetic diversity of Candida albicans in patients with vulvovaginal candidiasis
WANG Zhiheng, YING Chunmei, ZHAO Hu. (Department of Lobratory Medicine, Huadong Hospital Affliated to Fudan University, Shanghai 200040, China)
Abstract:Objective To investigate the molecular epidemiology of 114 C. albicans strains isolated from the vaginal discharge of female patients treated in three obstetrics and gynecology hospitals in Shanghai by analyzing the relationship between the main genotypes and resistance profle, and the relationship between genetic diversity and cluster of C. albicans. Methods A total of 114 strains of C. albicans were collected from the Obstetrics & Gynecology Hospital of Fudan University, Shanghai First Maternity and Infant Hospital Corporation and the International Peace Maternity & Child Health Hospital of China welfare institute. Phylogenetic analysis of strains were carried out by eBURST. C. albicans strains were also analyzed by multilocus sequence typing (MLST). The susceptibility of the C. albicans strains was tested by ATB FUNGUS 3. Results A total of 47 diploid strain types (DSTs) were identifed from the 114 strains, 30 of which were known types. DST 79 and DST 435 were the main types. Of the 114 C. albicans strains, 96.5% were susceptible to fucytosine, 100% to amphotericin B, 85.1% to fuconazole, 55.2% to itraconazole and 84.3% to voriconazole. Conclusions The pathogenic C. albicans strains isolated from different obstetrics and gynecology hospitals in Shanghai were originated from multiple clones, the main type of which was DST 79 and DST 435 with certain degree of antifungal resistance. MLST typing suggests that genetic diversity is present in the C. albicans strains isolated in Shanghai area. The clustering analysis of C. albicans strains is consistent with its genotypes.
Key words:C. albicans; multilocus sequence typing; genotyping
